Institute for Chemical Biology & Drug Discovery, Department of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA.
Biochemistry. 2013 Jun 18;52(24):4217-28. doi: 10.1021/bi400413c. Epub 2013 Jun 6.
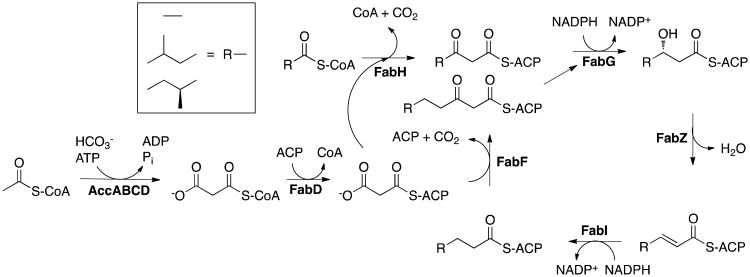
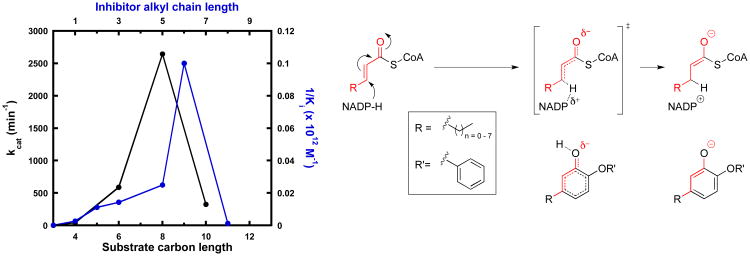
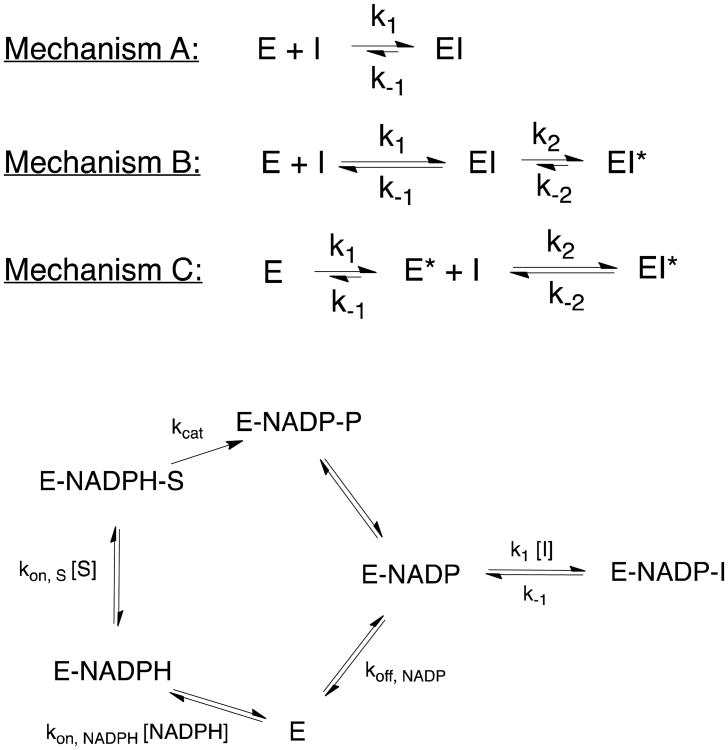
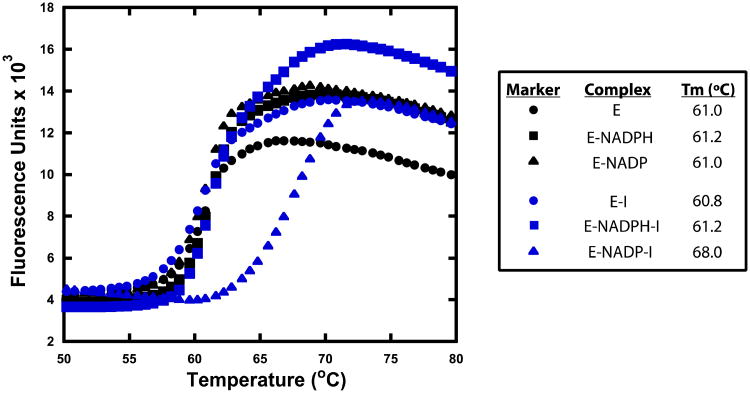
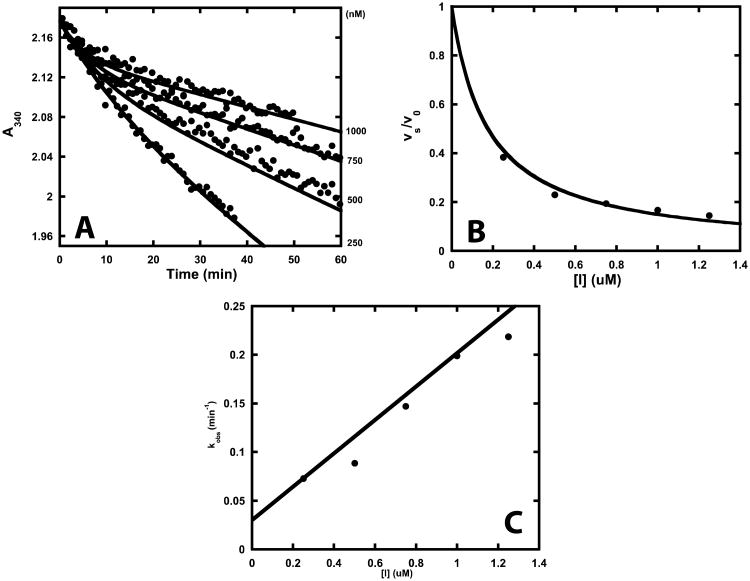
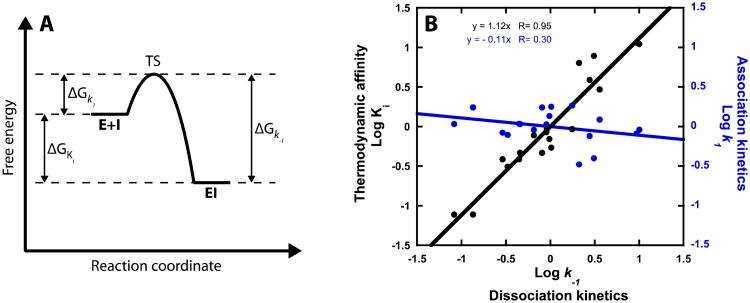
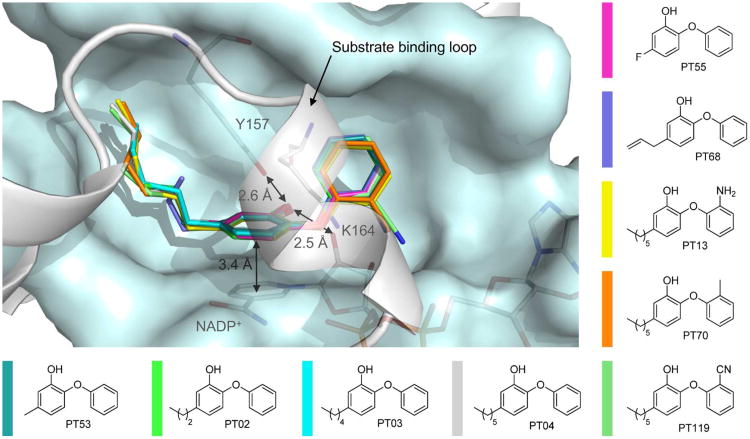
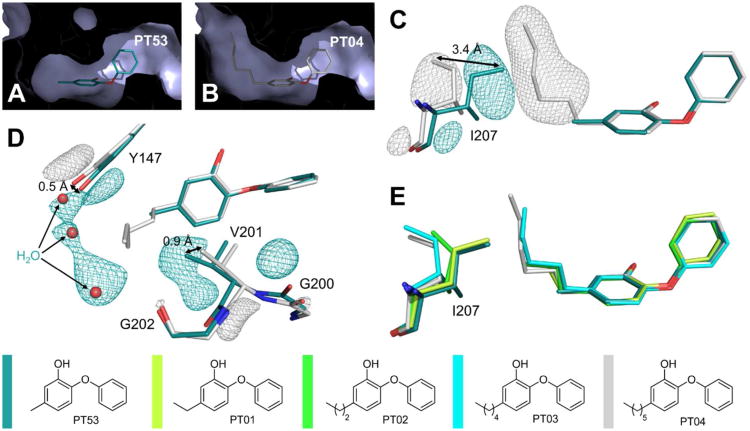
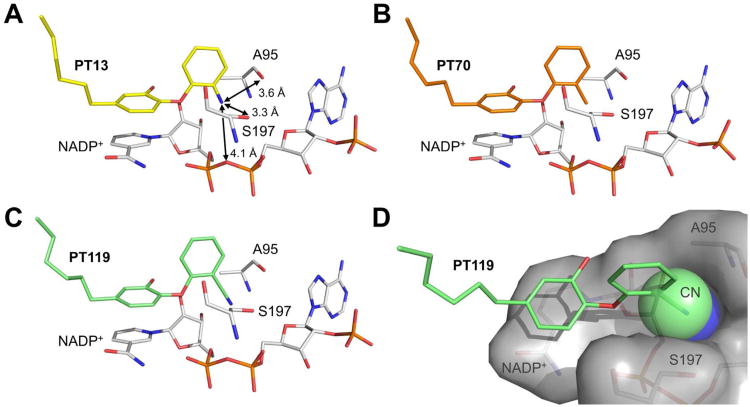
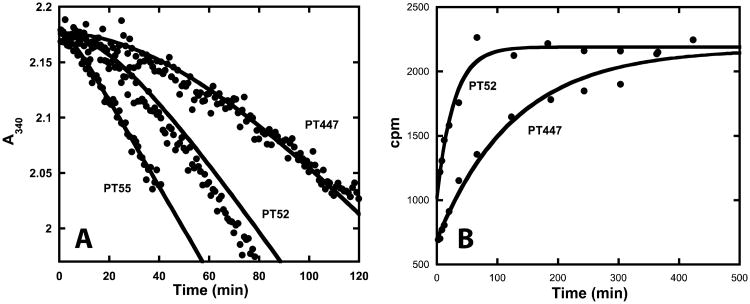
Drug-target kinetics has recently emerged as an especially important facet of the drug discovery process. In particular, prolonged drug-target residence times may confer enhanced efficacy and selectivity in the open in vivo system. However, the lack of accurate kinetic and structural data for a series of congeneric compounds hinders the rational design of inhibitors with decreased off-rates. Therefore, we chose the Staphylococcus aureus enoyl-ACP reductase (saFabI)--an important target for the development of new anti-staphylococcal drugs--as a model system to rationalize and optimize the drug-target residence time on a structural basis. Using our new, efficient, and widely applicable mechanistically informed kinetic approach, we obtained a full characterization of saFabI inhibition by a series of 20 diphenyl ethers complemented by a collection of 9 saFabI-inhibitor crystal structures. We identified a strong correlation between the affinities of the investigated saFabI diphenyl ether inhibitors and their corresponding residence times, which can be rationalized on a structural basis. Because of its favorable interactions with the enzyme, the residence time of our most potent compound exceeds 10 h. In addition, we found that affinity and residence time in this system can be significantly enhanced by modifications predictable by a careful consideration of catalysis. Our study provides a blueprint for investigating and prolonging drug-target kinetics and may aid in the rational design of long-residence-time inhibitors targeting the essential saFabI enzyme.
药物-靶标动力学最近成为药物发现过程中一个特别重要的方面。特别是,延长药物-靶标停留时间可以在开放的体内系统中赋予增强的疗效和选择性。然而,由于缺乏一系列同系物化合物的准确动力学和结构数据,因此难以合理设计降低脱靶率的抑制剂。因此,我们选择金黄色葡萄球菌烯酰-ACP 还原酶(saFabI)——开发新型抗葡萄球菌药物的重要靶标——作为模型系统,在结构基础上合理化和优化药物-靶标停留时间。我们使用新的、高效的、广泛适用的基于机制的信息动力学方法,对一系列 20 个二苯醚对 saFabI 的抑制作用进行了全面表征,并结合了 9 个 saFabI-抑制剂晶体结构的集合。我们确定了所研究的 saFabI 二苯醚抑制剂的亲和力与其相应停留时间之间存在很强的相关性,这可以在结构基础上得到合理化。由于与酶的有利相互作用,我们最有效的化合物的停留时间超过 10 小时。此外,我们发现,在该系统中,通过仔细考虑催化作用进行的修饰可以显著增强亲和力和停留时间。我们的研究为研究和延长药物-靶标动力学提供了蓝图,并可能有助于合理设计针对必需的 saFabI 酶的长停留时间抑制剂。