Department of Biostatistics, Erasmus MC, Rotterdam, The Netherlands.
BMC Bioinformatics. 2013 May 28;14:166. doi: 10.1186/1471-2105-14-166.
Genome-wide association studies have become very popular in identifying genetic contributions to phenotypes. Millions of SNPs are being tested for their association with diseases and traits using linear or logistic regression models. This conceptually simple strategy encounters the following computational issues: a large number of tests and very large genotype files (many Gigabytes) which cannot be directly loaded into the software memory. One of the solutions applied on a grand scale is cluster computing involving large-scale resources. We show how to speed up the computations using matrix operations in pure R code.
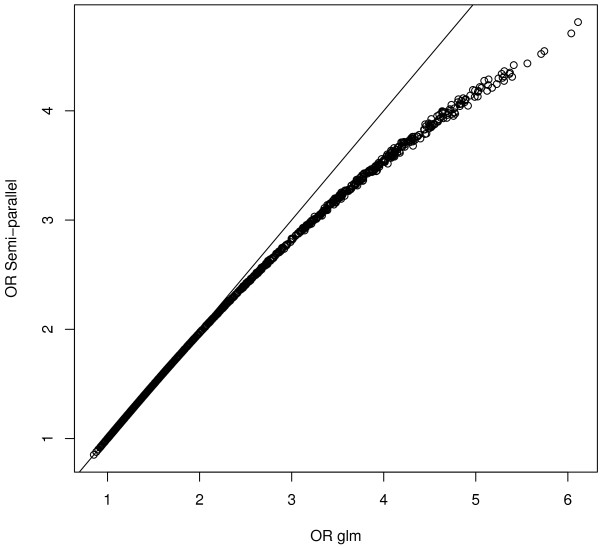
We improve speed: computation time from 6 hours is reduced to 10-15 minutes. Our approach can handle essentially an unlimited amount of covariates efficiently, using projections. Data files in GWAS are vast and reading them into computer memory becomes an important issue. However, much improvement can be made if the data is structured beforehand in a way allowing for easy access to blocks of SNPs. We propose several solutions based on the R packages ff and ncdf.We adapted the semi-parallel computations for logistic regression. We show that in a typical GWAS setting, where SNP effects are very small, we do not lose any precision and our computations are few hundreds times faster than standard procedures.
We provide very fast algorithms for GWAS written in pure R code. We also show how to rearrange SNP data for fast access.
全基因组关联研究已成为识别遗传对表型影响的热门方法。使用线性或逻辑回归模型,对数百万个 SNP 进行与疾病和特征的关联测试。这种概念上简单的策略遇到了以下计算问题:大量的测试和非常大的基因型文件(许多千兆字节),不能直接加载到软件内存中。一种大规模应用的解决方案是涉及大规模资源的集群计算。我们展示了如何使用纯 R 代码中的矩阵运算来加速计算。
我们提高了速度:计算时间从 6 小时缩短到 10-15 分钟。我们的方法可以有效地处理大量的协变量,使用投影。GWAS 中的数据文件非常庞大,将其读入计算机内存成为一个重要问题。但是,如果数据事先以允许轻松访问 SNP 块的方式进行结构化,就可以进行很大的改进。我们基于 R 包 ff 和 ncdf 提出了几种解决方案。我们对逻辑回归进行了半并行计算的改编。我们表明,在 SNP 效应非常小的典型 GWAS 环境中,我们不会损失任何精度,并且我们的计算速度比标准程序快几百倍。
我们提供了用纯 R 代码编写的非常快速的 GWAS 算法。我们还展示了如何重新排列 SNP 数据以快速访问。