Zheng X, Shen J, Cox C, Wakefield J C, Ehm M G, Nelson M R, Weir B S
Department of Biostatistics, University of Washington, Seattle, WA, USA.
Quantitative Sciences, GlaxoSmithKline, Research Triangle Park, NC, USA.
Pharmacogenomics J. 2014 Apr;14(2):192-200. doi: 10.1038/tpj.2013.18. Epub 2013 May 28.
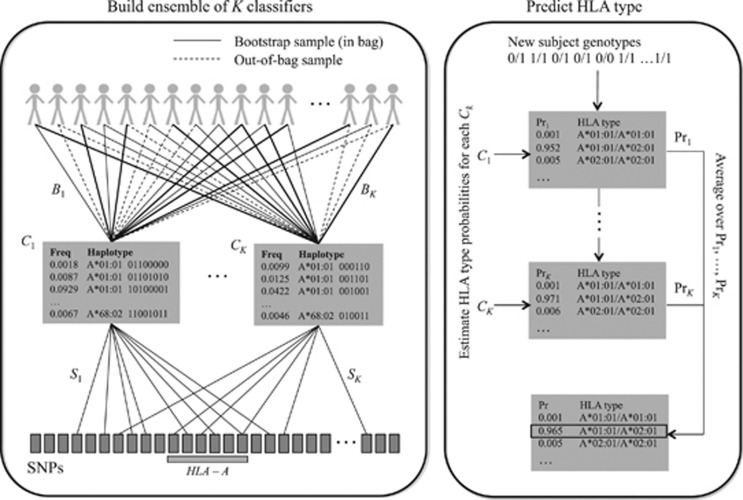
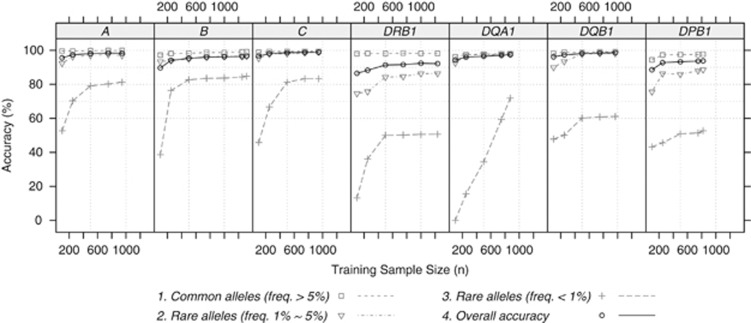
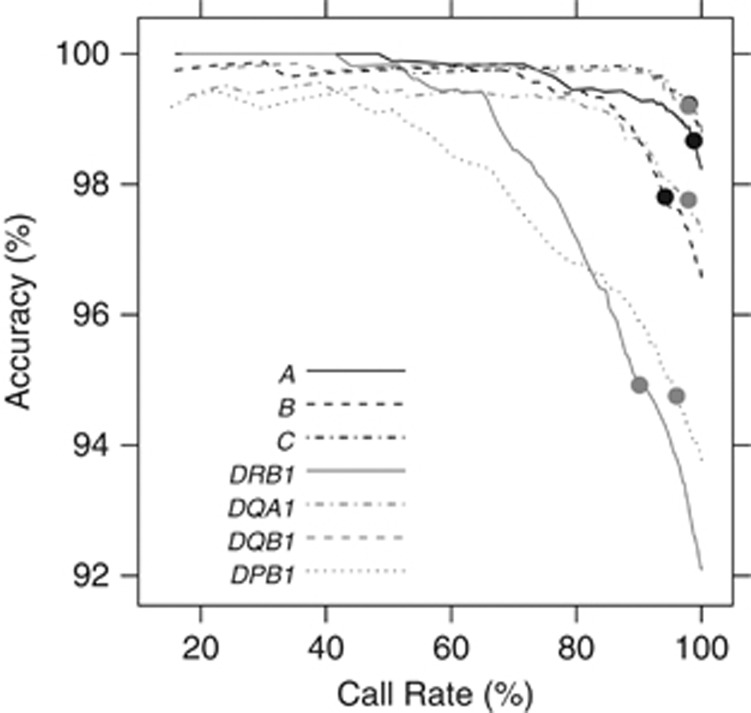
Genotyping of classical human leukocyte antigen (HLA) alleles is an essential tool in the analysis of diseases and adverse drug reactions with associations mapping to the major histocompatibility complex (MHC). However, deriving high-resolution HLA types subsequent to whole-genome single-nucleotide polymorphism (SNP) typing or sequencing is often cost prohibitive for large samples. An alternative approach takes advantage of the extended haplotype structure within the MHC to predict HLA alleles using dense SNP genotypes, such as those available from genome-wide SNP panels. Current methods for HLA imputation are difficult to apply or may require the user to have access to large training data sets with SNP and HLA types. We propose HIBAG, HLA Imputation using attribute BAGging, that makes predictions by averaging HLA-type posterior probabilities over an ensemble of classifiers built on bootstrap samples. We assess the performance of HIBAG using our study data (n=2668 subjects of European ancestry) as a training set and HLA data from the British 1958 birth cohort study (n≈1000 subjects) as independent validation samples. Prediction accuracies for HLA-A, B, C, DRB1 and DQB1 range from 92.2% to 98.1% using a set of SNP markers common to the Illumina 1M Duo, OmniQuad, OmniExpress, 660K and 550K platforms. HIBAG performed well compared with the other two leading methods, HLA*IMP and BEAGLE. This method is implemented in a freely available HIBAG R package that includes pre-fit classifiers for European, Asian, Hispanic and African ancestries, providing a readily available imputation approach without the need to have access to large training data sets.
经典人类白细胞抗原(HLA)等位基因的基因分型是分析与主要组织相容性复合体(MHC)相关的疾病和药物不良反应的重要工具。然而,在全基因组单核苷酸多态性(SNP)分型或测序之后获得高分辨率HLA类型对于大样本来说成本往往过高。一种替代方法利用MHC内扩展的单倍型结构,使用密集的SNP基因型(如全基因组SNP面板中的那些)来预测HLA等位基因。目前的HLA推断方法难以应用,或者可能要求用户能够获得具有SNP和HLA类型的大型训练数据集。我们提出了HIBAG,即使用属性装袋法进行HLA推断,它通过对基于自助抽样构建的分类器集合上的HLA类型后验概率求平均值来进行预测。我们使用我们的研究数据(n = 2668名欧洲血统受试者)作为训练集,并将来自英国1958年出生队列研究的HLA数据(n≈1000名受试者)作为独立验证样本,评估HIBAG的性能。使用Illumina 1M Duo、OmniQuad、OmniExpress、660K和550K平台共有的一组SNP标记,HLA - A、B、C、DRB1和DQB1的预测准确率在92.2%至98.1%之间。与其他两种领先方法HLA*IMP和BEAGLE相比,HIBAG表现良好。该方法在一个免费提供的HIBAG R包中实现,该包包括针对欧洲、亚洲、西班牙裔和非洲血统的预拟合分类器,提供了一种无需访问大型训练数据集即可随时使用的推断方法。