Department of Physiology and Biophysics,Weill Cornell Medical College, 1305 York Ave., New York, NY 10065, USA.
BMC Bioinformatics. 2013;14 Suppl 5(Suppl 5):S10. doi: 10.1186/1471-2105-14-S5-S10. Epub 2013 Apr 10.
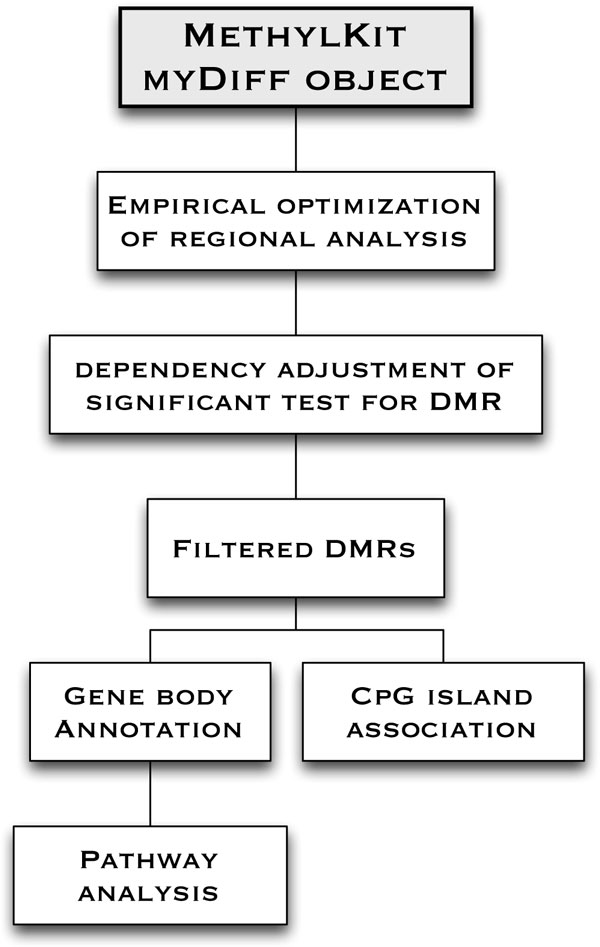
DNA methylation profiling reveals important differentially methylated regions (DMRs) of the genome that are altered during development or that are perturbed by disease. To date, few programs exist for regional analysis of enriched or whole-genome bisulfate conversion sequencing data, even though such data are increasingly common. Here, we describe an open-source, optimized method for determining empirically based DMRs (eDMR) from high-throughput sequence data that is applicable to enriched whole-genome methylation profiling datasets, as well as other globally enriched epigenetic modification data.
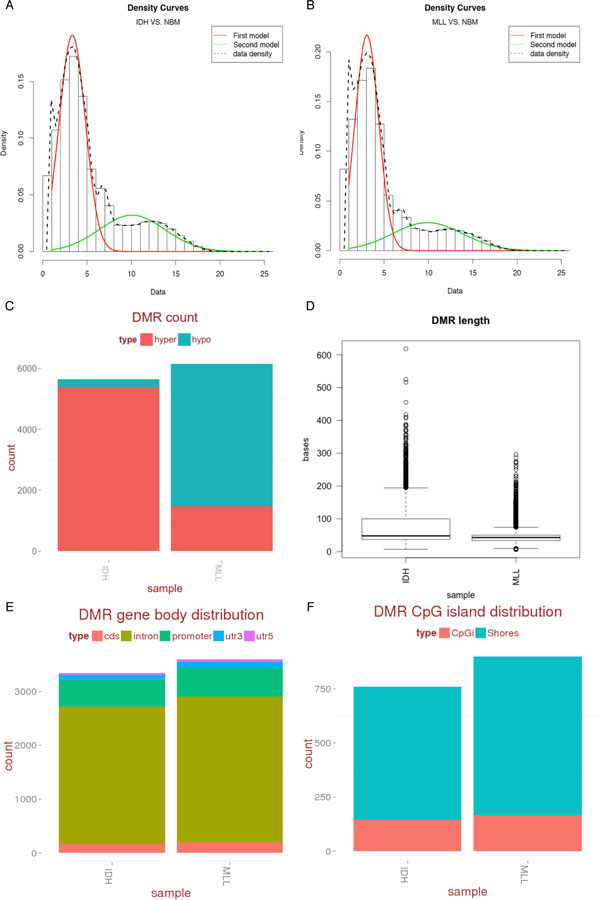
Here we show that our bimodal distribution model and weighted cost function for optimized regional methylation analysis provides accurate boundaries of regions harboring significant epigenetic modifications. Our algorithm takes the spatial distribution of CpGs into account for the enrichment assay, allowing for optimization of the definition of empirical regions for differential methylation. Combined with the dependent adjustment for regional p-value combination and DMR annotation, we provide a method that may be applied to a variety of datasets for rapid DMR analysis. Our method classifies both the directionality of DMRs and their genome-wide distribution, and we have observed that shows clinical relevance through correct stratification of two Acute Myeloid Leukemia (AML) tumor sub-types.
Our weighted optimization algorithm eDMR for calling DMRs extends an established DMR R pipeline (methylKit) and provides a needed resource in epigenomics. Our method enables an accurate and scalable way of finding DMRs in high-throughput methylation sequencing experiments. eDMR is available for download at http://code.google.com/p/edmr/.
DNA 甲基化分析揭示了在发育过程中发生改变或受疾病干扰的基因组中重要的差异甲基化区域(DMR)。迄今为止,尽管此类数据越来越常见,但用于富集或全基因组亚硫酸氢盐转换测序数据的区域分析的程序很少。在这里,我们描述了一种从高通量序列数据中确定基于经验的 DMR(eDMR)的开源、优化方法,该方法适用于富集的全基因组甲基化分析数据集以及其他全球富集的表观遗传修饰数据。
我们表明,我们的双峰分布模型和优化区域甲基化分析的加权成本函数为具有显著表观遗传修饰的区域提供了准确的边界。我们的算法考虑了 CpG 的空间分布,用于富集测定,允许对差异甲基化的经验区域进行优化定义。与依赖于区域 p 值组合和 DMR 注释的调整相结合,我们提供了一种可应用于各种数据集的快速 DMR 分析方法。我们的方法对 DMR 的方向和全基因组分布进行分类,并且我们已经观察到通过两种急性髓性白血病(AML)肿瘤亚型的正确分层来显示临床相关性。
我们用于调用 DMR 的基于加权优化的算法 eDMR 扩展了现有的 DMR R 管道(methylKit),并为表观基因组学提供了必要的资源。我们的方法提供了一种在高通量甲基化测序实验中寻找 DMR 的准确且可扩展的方法。eDMR 可在 http://code.google.com/p/edmr/ 下载。