ARC Centre of Excellence in Plant Energy Biology, The University of Western Australia, Perth, Australia.
Harry Perkins Institute of Medical Research, Perth, Australia.
BMC Bioinformatics. 2019 May 16;20(1):253. doi: 10.1186/s12859-019-2845-y.
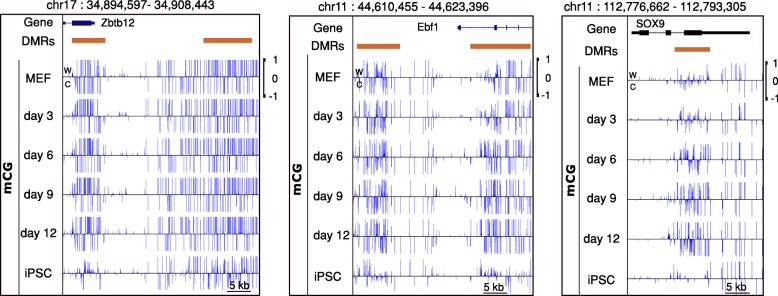
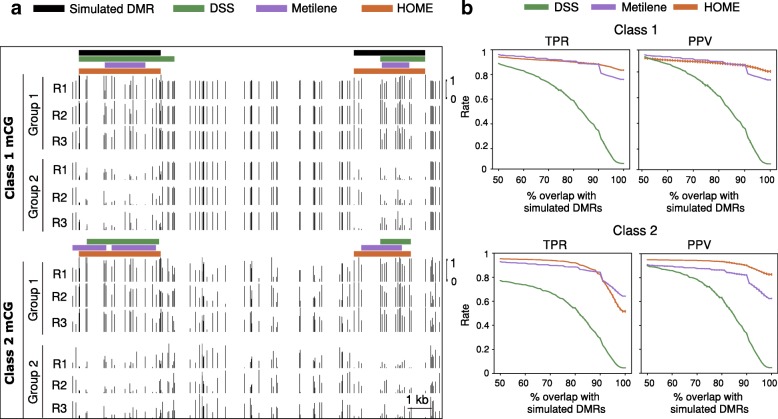
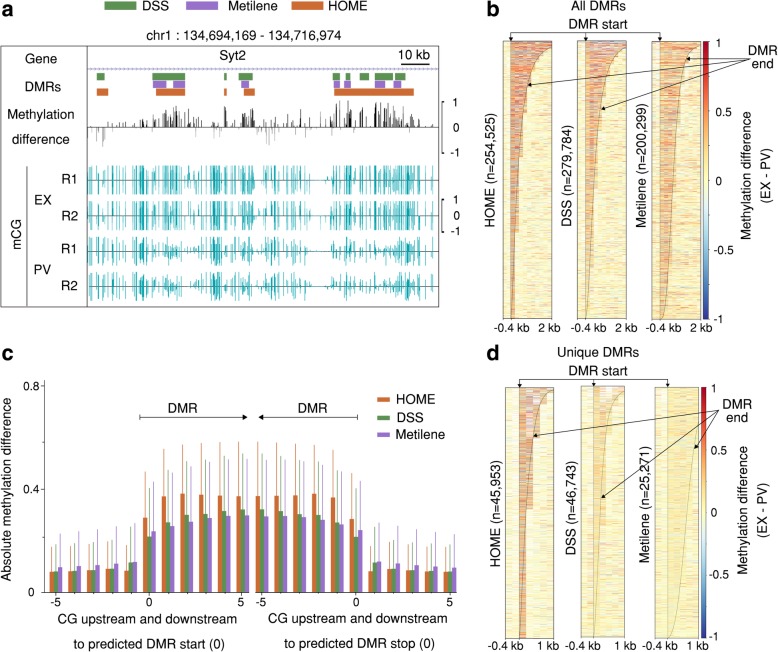
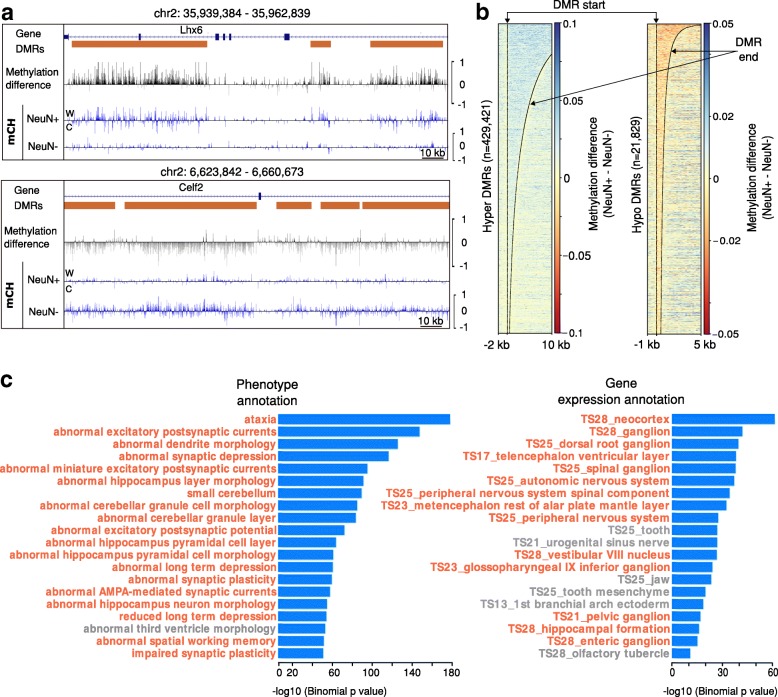
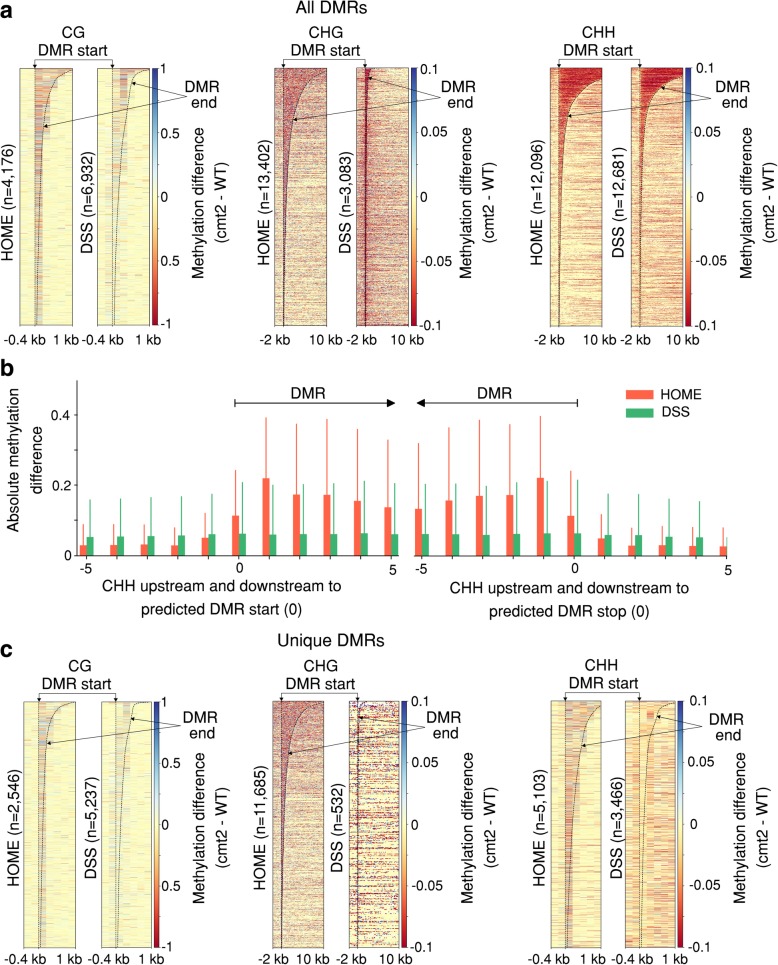
The development of whole genome bisulfite sequencing has made it possible to identify methylation differences at single base resolution throughout an entire genome. However, a persistent challenge in DNA methylome analysis is the accurate identification of differentially methylated regions (DMRs) between samples. Sensitive and specific identification of DMRs among different conditions requires accurate and efficient algorithms, and while various tools have been developed to tackle this problem, they frequently suffer from inaccurate DMR boundary identification and high false positive rate.
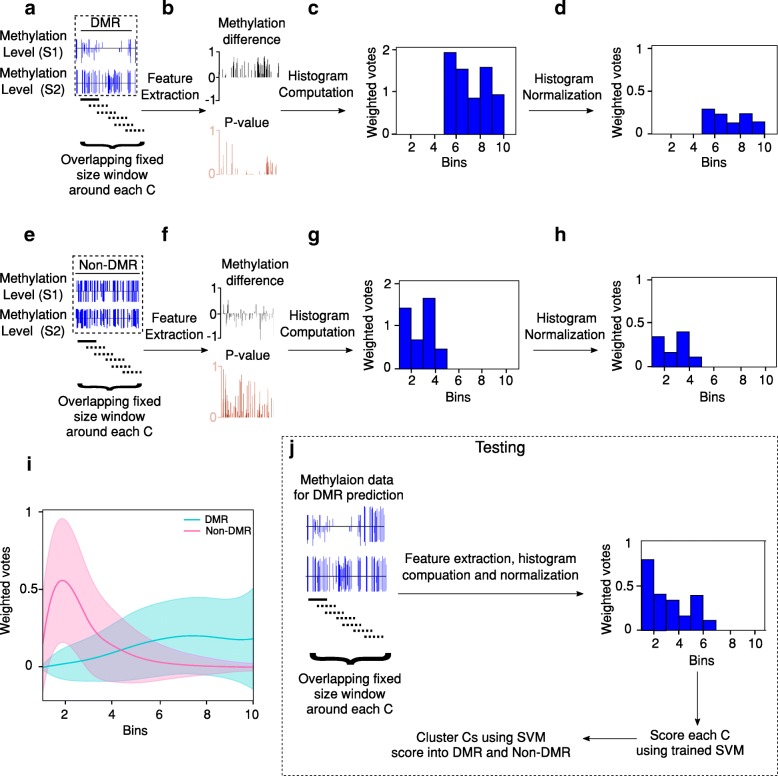
We present a novel Histogram Of MEthylation (HOME) based method that takes into account the inherent difference in the distribution of methylation levels between DMRs and non-DMRs to discriminate between the two using a Support Vector Machine. We show that generated features used by HOME are dataset-independent such that a classifier trained on, for example, a mouse methylome training set of regions of differentially accessible chromatin, can be applied to any other organism's dataset and identify accurate DMRs. We demonstrate that DMRs identified by HOME exhibit higher association with biologically relevant genes, processes, and regulatory events compared to the existing methods. Moreover, HOME provides additional functionalities lacking in most of the current DMR finders such as DMR identification in non-CG context and time series analysis. HOME is freely available at https://github.com/ListerLab/HOME .
HOME produces more accurate DMRs than the current state-of-the-art methods on both simulated and biological datasets. The broad applicability of HOME to identify accurate DMRs in genomic data from any organism will have a significant impact upon expanding our knowledge of how DNA methylation dynamics affect cell development and differentiation.
全基因组亚硫酸氢盐测序的发展使得在整个基因组中单碱基分辨率鉴定甲基化差异成为可能。然而,在 DNA 甲基化组分析中,一个持续存在的挑战是准确识别样品之间的差异甲基化区域(DMR)。在不同条件下敏感且特异性地识别 DMR 需要准确和高效的算法,尽管已经开发了各种工具来解决这个问题,但它们经常存在 DMR 边界识别不准确和高假阳性率的问题。
我们提出了一种新的基于Histogram Of MEthylation(HOME)的方法,该方法考虑了 DMR 和非 DMR 之间甲基化水平分布的固有差异,使用支持向量机对两者进行区分。我们表明,HOME 生成的特征与数据集无关,例如,在一个差异可及染色质区域的小鼠甲基化组训练集上训练的分类器可以应用于任何其他生物体的数据集,并准确识别 DMR。我们表明,与现有的方法相比,HOME 识别的 DMR 与生物相关基因、过程和调控事件的相关性更高。此外,HOME 提供了大多数当前 DMR 发现器所缺乏的附加功能,例如非 CG 背景下的 DMR 识别和时间序列分析。HOME 可在 https://github.com/ListerLab/HOME 上免费获得。
在模拟和生物数据集上,HOME 产生的 DMR 比当前最先进的方法更准确。HOME 广泛适用于从任何生物体中识别基因组数据中的准确 DMR,这将极大地促进我们对 DNA 甲基化动态如何影响细胞发育和分化的认识。