Laboratory of Emerging Pathogens, DETTD/OBRR/CBER, US Food and Drug Administration, Bethesda, Maryland, USA.
PLoS Negl Trop Dis. 2013 May 30;7(5):e2245. doi: 10.1371/journal.pntd.0002245. Print 2013.
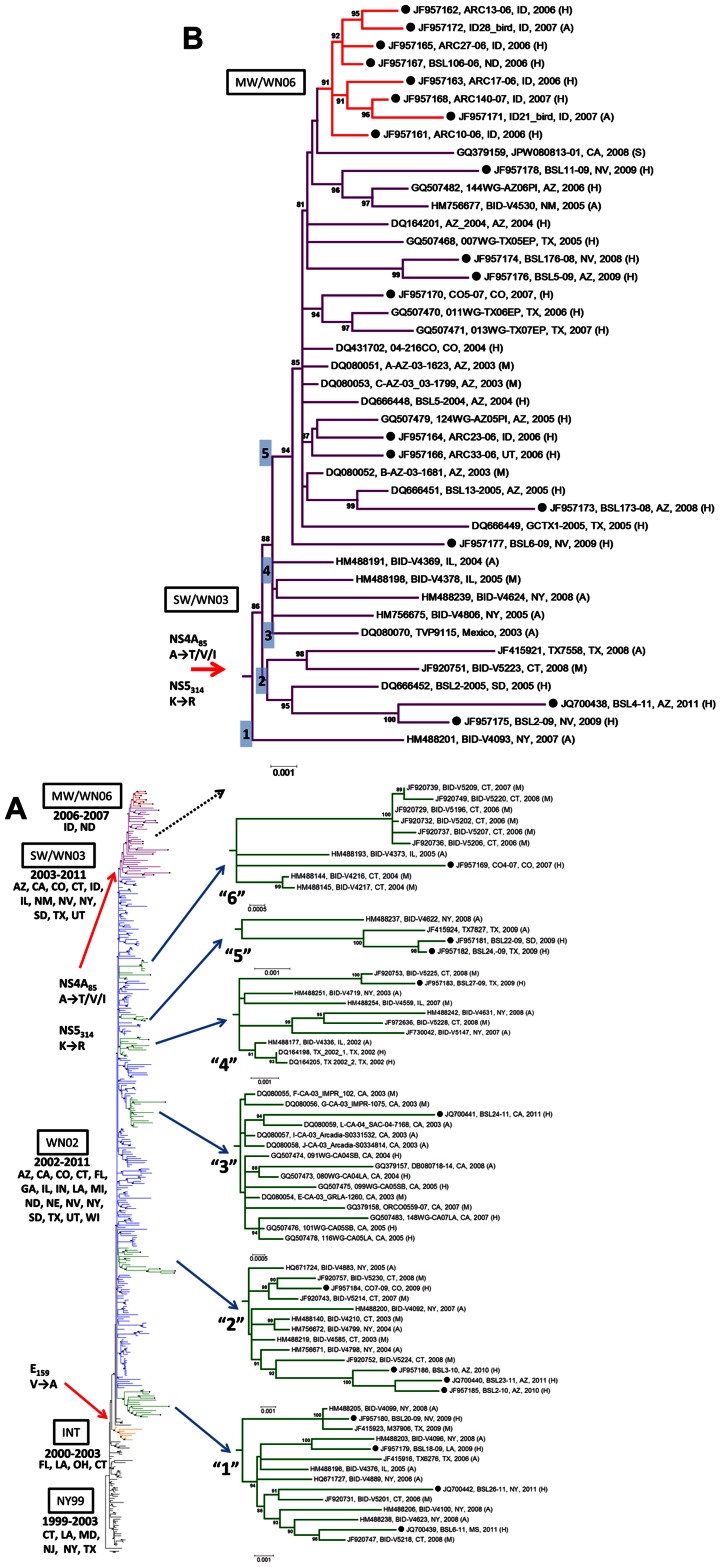
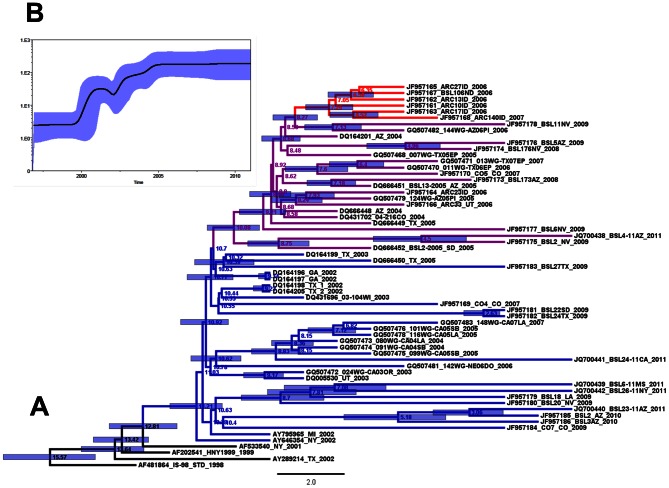
West Nile virus (WNV), an arbovirus maintained in a bird-mosquito enzootic cycle, can infect other vertebrates including humans. WNV was first reported in the US in 1999 where, to date, three genotypes belonging to WNV lineage I have been described (NY99, WN02, SW/WN03). We report here the WNV sequences obtained from two birds, one mosquito, and 29 selected human samples acquired during the US epidemics from 2006-2011 and our examination of the evolutionary dynamics in the open-reading frame of WNV isolates reported from 1999-2011. Maximum-likelihood and Bayesian methods were used to perform the phylogenetic analyses and selection pressure analyses were conducted with the HyPhy package. Phylogenetic analysis identified human WNV isolates within the main WNV genotypes that have circulated in the US. Within genotype SW/WN03, we have identified a cluster with strains derived from blood donors and birds from Idaho and North Dakota collected during 2006-2007, termed here MW/WN06. Using different codon-based and branch-site selection models, we detected a number of codons subjected to positive pressure in WNV genes. The mean nucleotide substitution rate for WNV isolates obtained from humans was calculated to be 5.06×10(-4) substitutions/site/year (s/s/y). The Bayesian skyline plot shows that after a period of high genetic variability following the introduction of WNV into the US, the WNV population appears to have reached genetic stability. The establishment of WNV in the US represents a unique opportunity to understand how an arbovirus adapts and evolves in a naïve environment. We describe a novel, well-supported cluster of WNV formed by strains collected from humans and birds from Idaho and North Dakota. Adequate genetic surveillance is essential to public health since new mutants could potentially affect viral pathogenesis, decrease performance of diagnostic assays, and negatively impact the efficacy of vaccines and the development of specific therapies.
西尼罗河病毒(WNV)是一种通过鸟类-蚊子媒介维持的虫媒病毒,可以感染包括人类在内的其他脊椎动物。WNV 于 1999 年在美国首次报告,迄今为止,已描述了属于 WNV 谱系 I 的三种基因型(NY99、WN02、SW/WN03)。我们在此报告了从 2006 年至 2011 年在美国流行期间获得的两只鸟类、一只蚊子和 29 个人类样本中获得的 WNV 序列,以及我们对 1999 年至 2011 年报告的 WNV 分离株开放阅读框进化动态的研究。最大似然法和贝叶斯法用于进行系统发育分析,HyPhy 包用于进行选择压力分析。系统发育分析确定了在美国流行的主要 WNV 基因型中的人类 WNV 分离株。在基因型 SW/WN03 内,我们鉴定出了一个聚类,其中包括来自 2006 年至 2007 年期间来自爱达荷州和北达科他州的献血者和鸟类来源的菌株,我们将其命名为 MW/WN06。使用不同的基于密码子和分支位点选择模型,我们检测到 WNV 基因中存在多个受正选择压力影响的密码子。从人类中获得的 WNV 分离株的平均核苷酸取代率计算为 5.06×10(-4) 个替换/位点/年(s/s/y)。贝叶斯天空线图显示,WNV 在美国传入后经历了一段时间的高遗传变异性,WNV 种群似乎已经达到遗传稳定。WNV 在 美国的建立为了解一种虫媒病毒如何在新环境中适应和进化提供了一个独特的机会。我们描述了一个由来自爱达荷州和北达科他州的人类和鸟类样本中收集的菌株组成的新型、有充分支持的 WNV 聚类。充分的遗传监测对于公共卫生至关重要,因为新的突变株可能会影响病毒的发病机制,降低诊断检测的性能,并对疫苗的疗效和特定疗法的发展产生负面影响。