Manchester Centre for Genomic Medicine, Institute of Human Development, University of Manchester, Manchester, UK.
Orphanet J Rare Dis. 2013 Jun 13;8:84. doi: 10.1186/1750-1172-8-84.
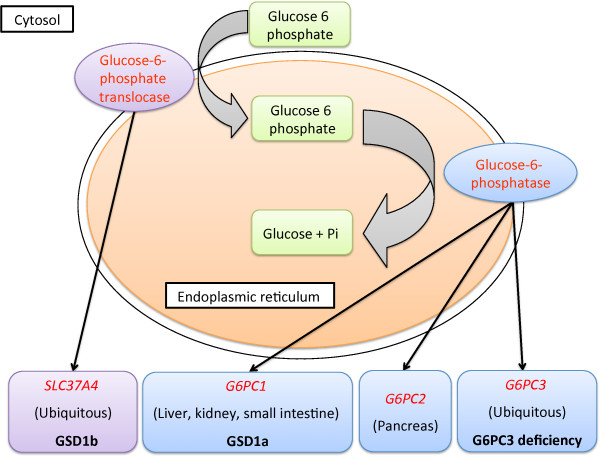
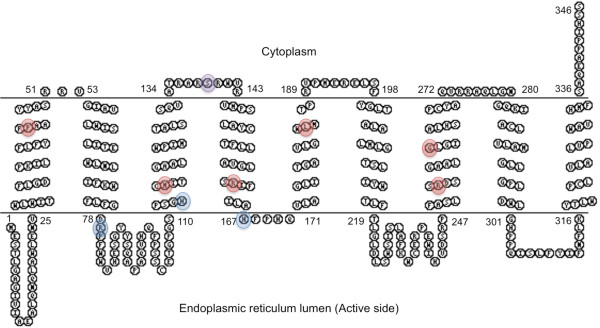
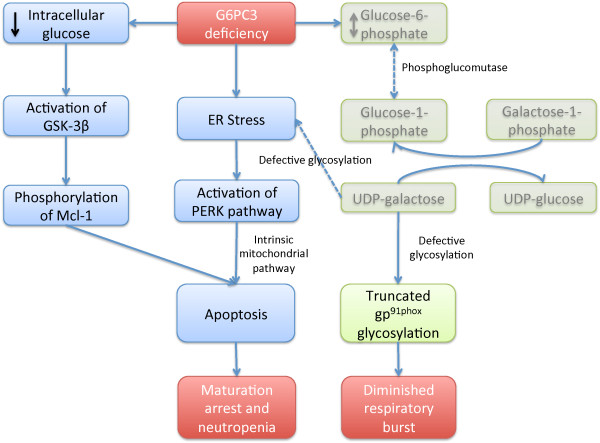
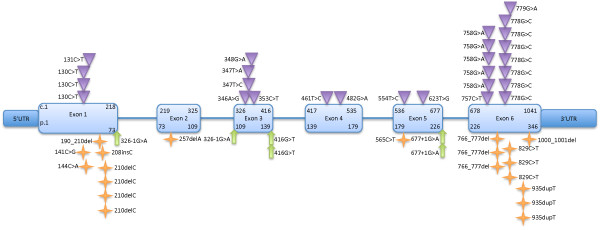
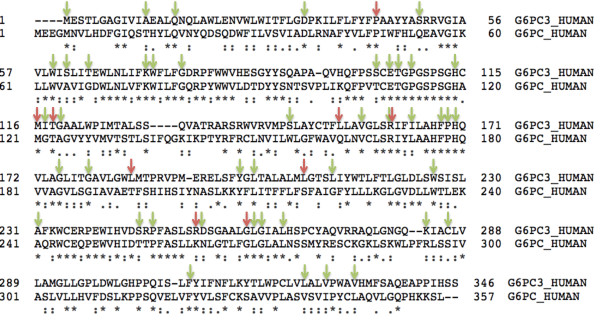
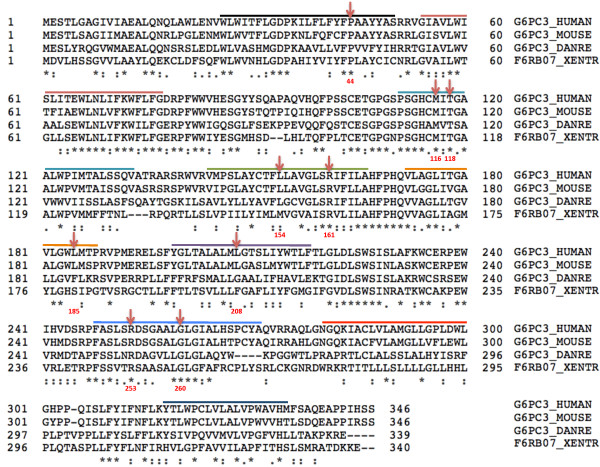
The G6PC3 gene encodes the ubiquitously expressed glucose-6-phosphatase enzyme (G-6-Pase β or G-6-Pase 3 or G6PC3). Bi-allelic G6PC3 mutations cause a multi-system autosomal recessive disorder of G6PC3 deficiency (also called severe congenital neutropenia type 4, MIM 612541). To date, at least 57 patients with G6PC3 deficiency have been described in the literature.G6PC3 deficiency is characterized by severe congenital neutropenia, recurrent bacterial infections, intermittent thrombocytopenia in many patients, a prominent superficial venous pattern and a high incidence of congenital cardiac defects and uro-genital anomalies. The phenotypic spectrum of the condition is wide and includes rare manifestations such as maturation arrest of the myeloid lineage, a normocellular bone marrow, myelokathexis, lymphopaenia, thymic hypoplasia, inflammatory bowel disease, primary pulmonary hypertension, endocrine abnormalities, growth retardation, minor facial dysmorphism, skeletal and integument anomalies amongst others. Dursun syndrome is part of this extended spectrum. G6PC3 deficiency can also result in isolated non-syndromic severe neutropenia. G6PC3 mutations in result in reduced enzyme activity, endoplasmic reticulum stress response, increased rates of apoptosis of affected cells and dysfunction of neutrophil activity.In this review we demonstrate that loss of function in missense G6PC3 mutations likely results from decreased enzyme stability. The condition can be diagnosed by sequencing the G6PC3 gene. A number of G6PC3 founder mutations are known in various populations and a possible genotype-phenotype relationship also exists. G6PC3 deficiency should be considered as part of the differential diagnoses in any patient with unexplained congenital neutropenia.Treatment with G-CSF leads to improvement in neutrophil numbers, prevents infections and improves quality of life. Mildly affected patients can be managed with prophylactic antibiotics. Untreated G6PC3 deficiency can be fatal. Echocardiogram, renal and pelvic ultrasound scans should be performed in all cases of suspected or confirmed G6PC3 deficiency. Routine assessment should include biochemical profile, growth profile and monitoring for development of varicose veins or venous ulcers.
G6PC3 基因编码广泛表达的葡萄糖-6-磷酸酶(G-6-Pase β 或 G-6-Pase 3 或 G6PC3)。双等位基因 G6PC3 突变导致 G6PC3 缺乏的多系统常染色体隐性遗传疾病(也称为严重先天性中性粒细胞减少症 4,MIM 612541)。迄今为止,文献中已描述了至少 57 例 G6PC3 缺乏症患者。G6PC3 缺乏症的特征是严重先天性中性粒细胞减少症、反复细菌感染、许多患者间歇性血小板减少症、明显的浅表静脉模式以及先天性心脏缺陷和泌尿生殖系统异常的高发率。该病症的表型谱很广,包括骨髓髓系成熟停滞、骨髓细胞正常、骨髓化生、淋巴细胞减少症、胸腺发育不全、炎症性肠病、原发性肺动脉高压、内分泌异常、生长迟缓、轻度面部畸形、骨骼和皮肤等罕见表现。Dursun 综合征是该扩展谱的一部分。G6PC3 缺乏症也可导致孤立的非综合征性严重中性粒细胞减少症。G6PC3 突变导致酶活性降低、内质网应激反应、受影响细胞凋亡率增加和中性粒细胞功能障碍。在本综述中,我们证明错义 G6PC3 突变的功能丧失可能是由于酶稳定性降低所致。可以通过测序 G6PC3 基因来诊断该疾病。在不同人群中已知存在许多 G6PC3 启动子突变,也存在可能的基因型-表型关系。在任何不明原因的先天性中性粒细胞减少症患者中,都应考虑 G6PC3 缺乏症作为鉴别诊断的一部分。用 G-CSF 治疗可改善中性粒细胞数量,预防感染并提高生活质量。轻度受影响的患者可以预防性使用抗生素治疗。未经治疗的 G6PC3 缺乏症可能致命。在疑似或确诊 G6PC3 缺乏症的所有病例中,应进行超声心动图、肾和骨盆超声扫描。常规评估应包括生化谱、生长谱以及监测静脉曲张或静脉溃疡的发展。