Zhen Xin, Betti Michael J, Kars Meltem Ece, Patterson Andrew R, Medina-Torres Edgar Alejandro, Scheffler Mendoza Selma Cecilia, Herrera Sánchez Diana Andrea, Lopez-Herrera Gabriela, Svyryd Yevgeniya, Mutchinick Osvaldo M, Gamazon Eric R, Rathmell Jeffrey C, Itan Yuval, Markle Janet, O'Farrill Romanillos Patricia, Lugo-Reyes Saul Oswaldo, Martinez-Barricarte Ruben
Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA.
Division of Molecular Pathogenesis, Department of Pathology Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, USA.
J Clin Immunol. 2024 Dec 4;45(1):53. doi: 10.1007/s10875-024-01836-0.
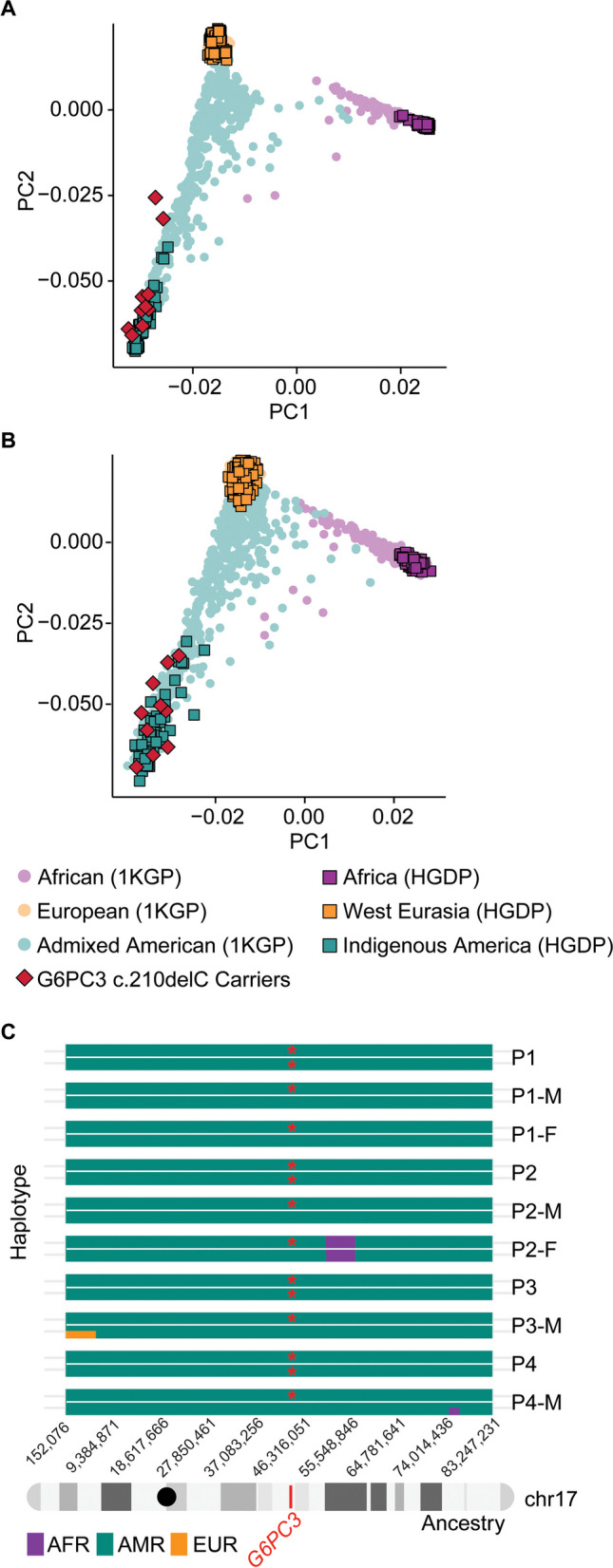
G6PC3 deficiency is a monogenic immunometabolic disorder that causes severe congenital neutropenia type 4. Patients display heterogeneous extra-hematological manifestations, contributing to delayed diagnosis. Here, we investigated the origin and functional consequence of the G6PC3 c.210delC variant found in patients of Mexican descent. Based on the shared haplotypes amongst mutation carriers, we estimated that this variant originated from a founder effect in a common ancestor. Furthermore, by ancestry analysis, we concluded that it appeared in the indigenous Mexican population. At the protein level, we showed that this frameshift mutation leads to an aberrant protein expression in overexpression and patient-derived Epstein-Barr Virus-immortalized B (EBV-B) cells. The neutropenia observed in G6PC3-deficient patients is driven by the intracellular accumulation of the metabolite 1,5-anhydroglucitol-6-phosphate (1,5-AG6P) that inhibits glycolysis. We characterized how the c.210delC variant impacts glycolysis by performing extracellular flux assays on patient-derived EBV-B cells. When treated with 1,5-anhydroglucitol (1,5-AG), the precursor to 1,5-AG6P, patient cells exhibited markedly reduced engagement of glycolysis. Finally, we compared the clinical presentation of patients with the mutation c.210delC and all other G6PC3-deficient patients reported in the literature, and we found that the c.210delC carriers display all prominent clinical features observed in prior patients. In conclusion, G6PC3 c.210delC is a loss-of-function mutation that arose from a founder effect in the indigenous Mexican population. These findings may facilitate the diagnosis of additional patients in this geographical area. Moreover, the in vitro 1,5-AG-dependent functional assay used in our study could be employed to assess the pathogenicity of additional G6PC3 variants.
G6PC3缺乏症是一种单基因免疫代谢紊乱疾病,可导致严重的4型先天性中性粒细胞减少症。患者表现出异质性的血液外表现,导致诊断延迟。在此,我们研究了在墨西哥裔患者中发现的G6PC3 c.210delC变异的起源和功能后果。基于突变携带者之间共享的单倍型,我们估计该变异起源于一个共同祖先的奠基者效应。此外,通过血统分析,我们得出结论,它出现在墨西哥本土人群中。在蛋白质水平上,我们表明这种移码突变导致在过表达和患者来源的爱泼斯坦-巴尔病毒永生化B(EBV-B)细胞中出现异常的蛋白质表达。G6PC3缺乏症患者中观察到的中性粒细胞减少是由抑制糖酵解的代谢物1,5-脱水葡萄糖醇-6-磷酸(1,5-AG6P)在细胞内积累所驱动的。我们通过对患者来源的EBV-B细胞进行细胞外通量分析,表征了c.210delC变异如何影响糖酵解。当用1,5-AG6P的前体1,5-脱水葡萄糖醇(1,5-AG)处理时,患者细胞的糖酵解参与明显减少。最后,我们比较了携带c.210delC突变的患者与文献中报道的所有其他G6PC3缺乏症患者的临床表现,我们发现c.210delC携带者表现出先前患者中观察到的所有突出临床特征。总之,G6PC3 c.210delC是一种功能丧失突变,起源于墨西哥本土人群的奠基者效应。这些发现可能有助于该地理区域其他患者的诊断。此外,我们研究中使用的体外1,5-AG依赖性功能测定可用于评估其他G6PC3变异体的致病性。