Center for Computer Vision and Department of Mathematics, Sun Yat-Sen University, Guangzhou, China.
PLoS One. 2013 Jun 17;8(6):e66256. doi: 10.1371/journal.pone.0066256. Print 2013.
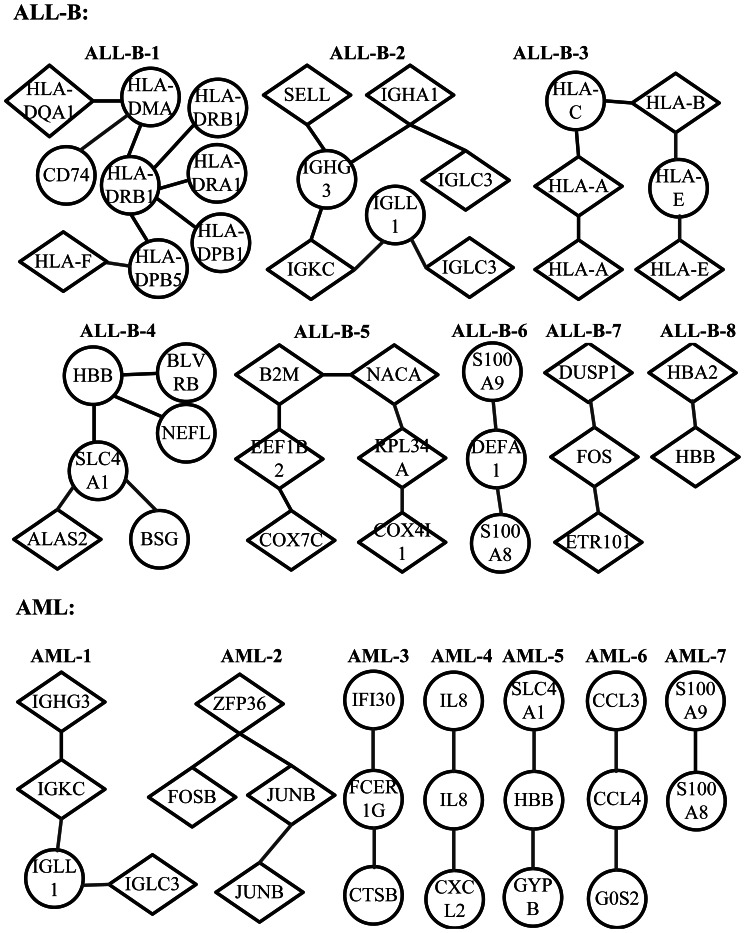
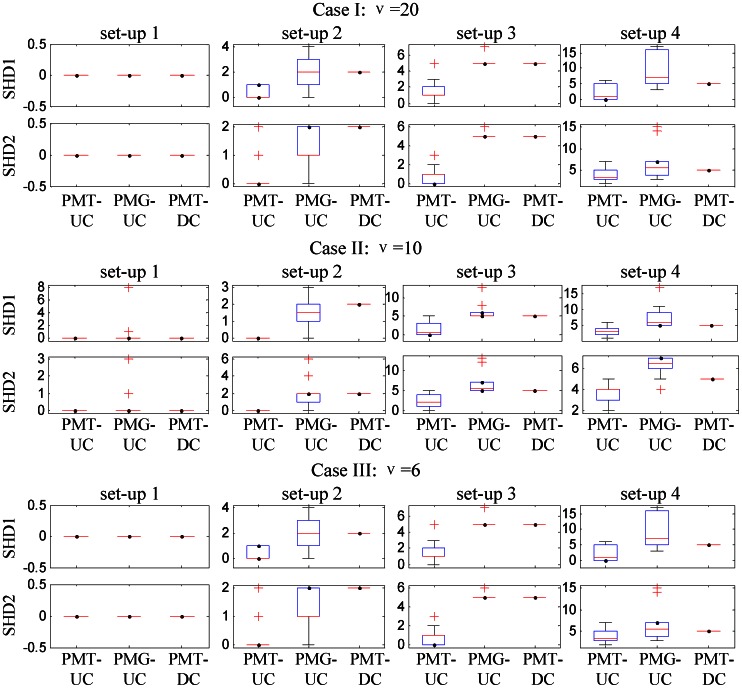
In cancer biology, it is very important to understand the phenotypic changes of the patients and discover new cancer subtypes. Recently, microarray-based technologies have shed light on this problem based on gene expression profiles which may contain outliers due to either chemical or electrical reasons. These undiscovered subtypes may be heterogeneous with respect to underlying networks or pathways, and are related with only a few of interdependent biomarkers. This motivates a need for the robust gene expression-based methods capable of discovering such subtypes, elucidating the corresponding network structures and identifying cancer related biomarkers. This study proposes a penalized model-based Student's t clustering with unconstrained covariance (PMT-UC) to discover cancer subtypes with cluster-specific networks, taking gene dependencies into account and having robustness against outliers. Meanwhile, biomarker identification and network reconstruction are achieved by imposing an adaptive [Formula: see text] penalty on the means and the inverse scale matrices. The model is fitted via the expectation maximization algorithm utilizing the graphical lasso. Here, a network-based gene selection criterion that identifies biomarkers not as individual genes but as subnetworks is applied. This allows us to implicate low discriminative biomarkers which play a central role in the subnetwork by interconnecting many differentially expressed genes, or have cluster-specific underlying network structures. Experiment results on simulated datasets and one available cancer dataset attest to the effectiveness, robustness of PMT-UC in cancer subtype discovering. Moveover, PMT-UC has the ability to select cancer related biomarkers which have been verified in biochemical or biomedical research and learn the biological significant correlation among genes.
在癌症生物学中,了解患者的表型变化并发现新的癌症亚型非常重要。最近,基于微阵列的技术根据基因表达谱揭示了这个问题,这些基因表达谱可能由于化学或电气原因而包含异常值。这些未被发现的亚型在潜在的网络或途径方面可能是异构的,并且仅与少数相互依赖的生物标志物相关。这就需要有稳健的基于基因表达的方法来发现这些亚型,阐明相应的网络结构,并识别癌症相关的生物标志物。本研究提出了一种基于惩罚模型的学生 t 聚类方法,该方法具有无约束协方差(PMT-UC),可以发现具有特定网络的癌症亚型,同时考虑基因依赖性,并具有对异常值的鲁棒性。同时,通过对均值和逆尺度矩阵施加自适应[Formula: see text]惩罚,实现了生物标志物的识别和网络重构。该模型通过利用图形套索的期望最大化算法进行拟合。这里,应用了一种基于网络的基因选择标准,该标准不是将生物标志物识别为单个基因,而是将其识别为子网络,从而可以确定在子网络中起中心作用的低判别性生物标志物,这些生物标志物通过连接许多差异表达的基因,或者具有特定于子网络的潜在网络结构。在模拟数据集和一个可用的癌症数据集上的实验结果证明了 PMT-UC 在癌症亚型发现中的有效性和鲁棒性。此外,PMT-UC 还具有选择已在生化或生物医学研究中得到验证的癌症相关生物标志物的能力,并学习基因之间具有生物学意义的相关性。