Center for Systems & Synthetic Biology, Institute for Cellular & Molecular Biology, The University of Texas at Austin, Austin, TX 78712, USA.
BMC Bioinformatics. 2013 Jun 21;14:203. doi: 10.1186/1471-2105-14-203.
Phenotypes and diseases may be related to seemingly dissimilar phenotypes in other species by means of the orthology of underlying genes. Such "orthologous phenotypes," or "phenologs," are examples of deep homology, and may be used to predict additional candidate disease genes.
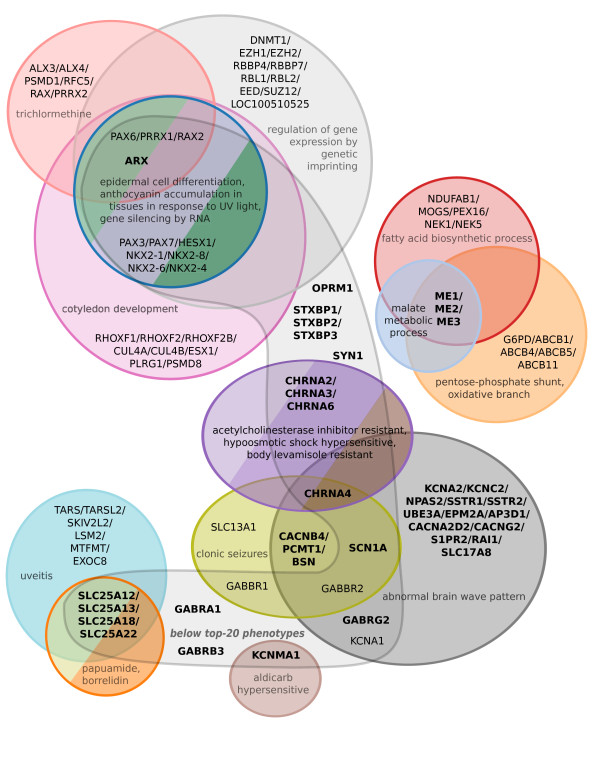
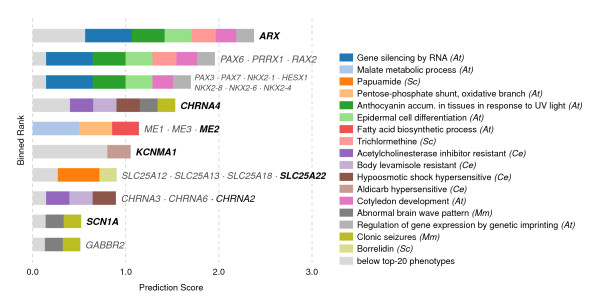
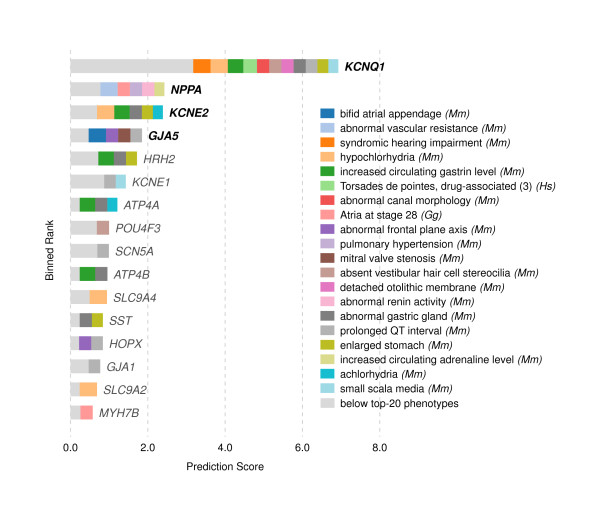
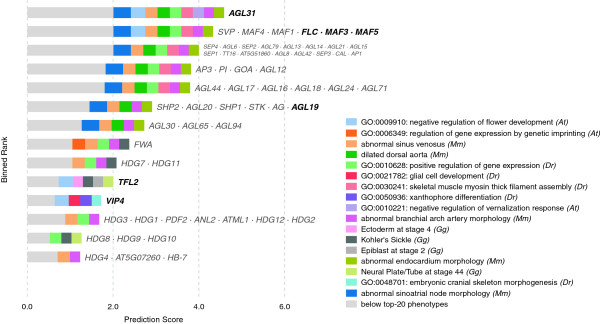
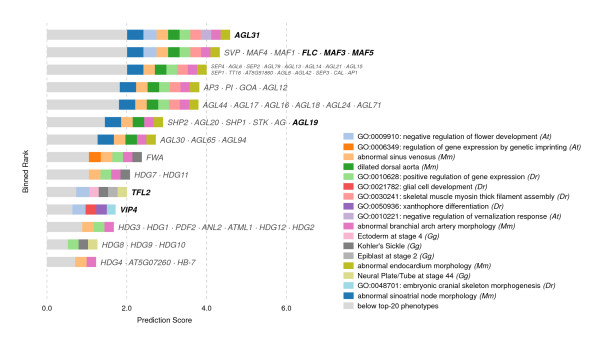
In this work, we develop an unsupervised algorithm for ranking phenolog-based candidate disease genes through the integration of predictions from the k nearest neighbor phenologs, comparing classifiers and weighting functions by cross-validation. We also improve upon the original method by extending the theory to paralogous phenotypes. Our algorithm makes use of additional phenotype data--from chicken, zebrafish, and E. coli, as well as new datasets for C. elegans--establishing that several types of annotations may be treated as phenotypes. We demonstrate the use of our algorithm to predict novel candidate genes for human atrial fibrillation (such as HRH2, ATP4A, ATP4B, and HOPX) and epilepsy (e.g., PAX6 and NKX2-1). We suggest gene candidates for pharmacologically-induced seizures in mouse, solely based on orthologous phenotypes from E. coli. We also explore the prediction of plant gene-phenotype associations, as for the Arabidopsis response to vernalization phenotype.
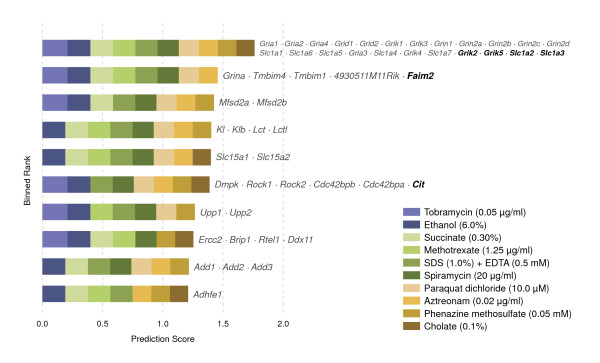
We are able to rank gene predictions for a significant portion of the diseases in the Online Mendelian Inheritance in Man database. Additionally, our method suggests candidate genes for mammalian seizures based only on bacterial phenotypes and gene orthology. We demonstrate that phenotype information may come from diverse sources, including drug sensitivities, gene ontology biological processes, and in situ hybridization annotations. Finally, we offer testable candidates for a variety of human diseases, plant traits, and other classes of phenotypes across a wide array of species.
通过潜在基因的同源性,表型和疾病可能与其他物种中看似不同的表型有关。这种“同源表型”或“表型同源物”是深度同源性的例子,可用于预测其他候选疾病基因。
在这项工作中,我们开发了一种无监督的算法,通过整合来自最近邻表型的预测,通过交叉验证比较分类器和加权函数,对基于表型的候选疾病基因进行排名。我们还通过将该理论扩展到旁系同源物来改进原始方法。我们的算法利用了来自鸡、斑马鱼和大肠杆菌的其他表型数据以及秀丽隐杆线虫的新数据集,从而可以将几种类型的注释视为表型。我们证明了我们的算法可用于预测人类心房颤动(如 HRH2、ATP4A、ATP4B 和 HOPX)和癫痫(例如 PAX6 和 NKX2-1)的新候选基因。我们仅根据大肠杆菌的同源表型,就提出了用于小鼠药物诱导性癫痫的候选基因。我们还探讨了植物基因-表型关联的预测,例如拟南芥对春化表型的反应。
我们能够对在线孟德尔遗传数据库中很大一部分疾病的基因预测进行排名。此外,我们的方法仅基于细菌表型和基因同源性,就提出了哺乳动物癫痫的候选基因。我们证明表型信息可以来自多种来源,包括药物敏感性、基因本体生物学过程和原位杂交注释。最后,我们为多种人类疾病、植物性状和其他广泛物种的表型类别的提供了可测试的候选基因。