Starke Sven, Meinke Peter, Camozzi Daria, Mattioli Elisabetta, Pfaeffle Roland, Siekmeyer Manuela, Hirsch Wolfgang, Horn Lars Christian, Paasch Uwe, Mitter Diana, Lattanzi Giovanna, Wehnert Manfred, Kiess Wieland
Department of Women and Child Health, Hospital for Children and Adolescents, Centre of Pediatric Research, University Hospital, University of Leipzig, Leipzig, Germany.
Aging (Albany NY). 2013 Jun;5(6):445-59. doi: 10.18632/aging.100566.
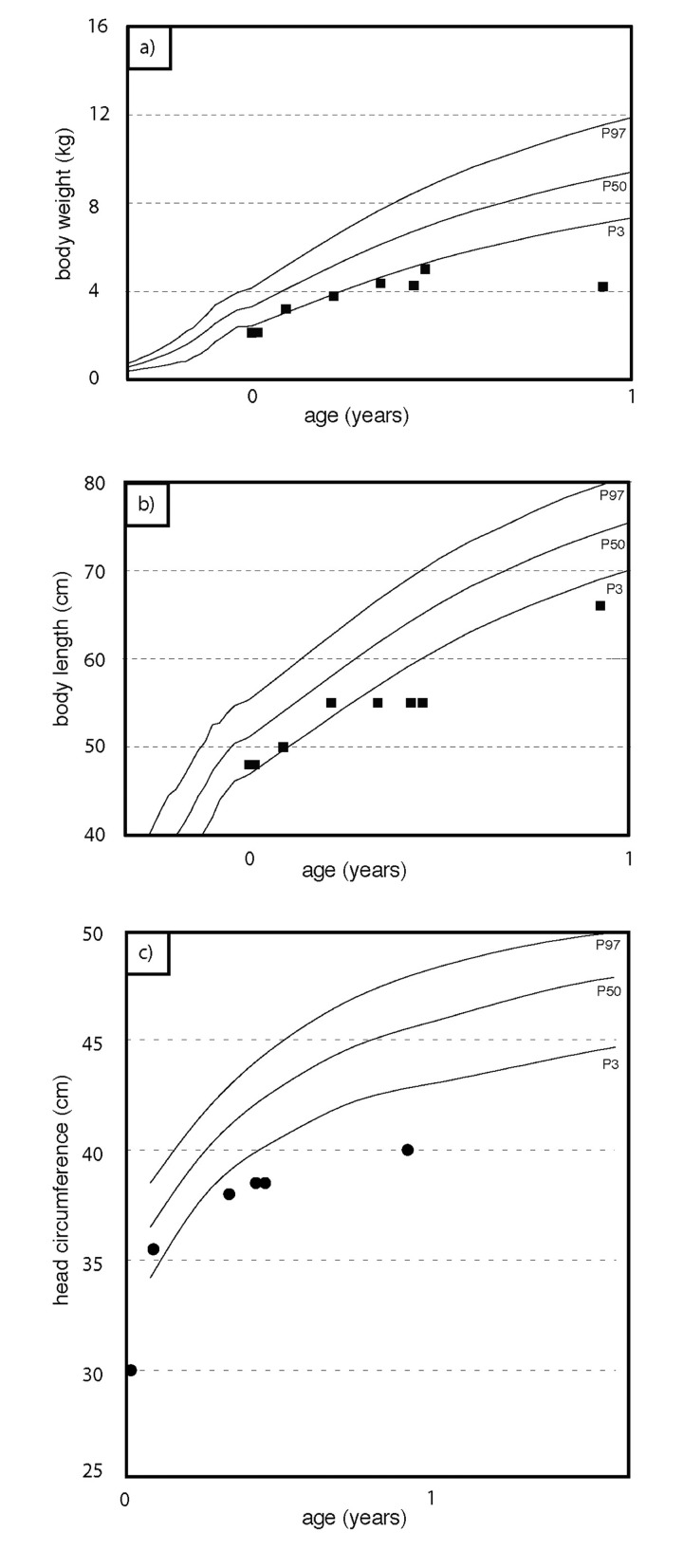
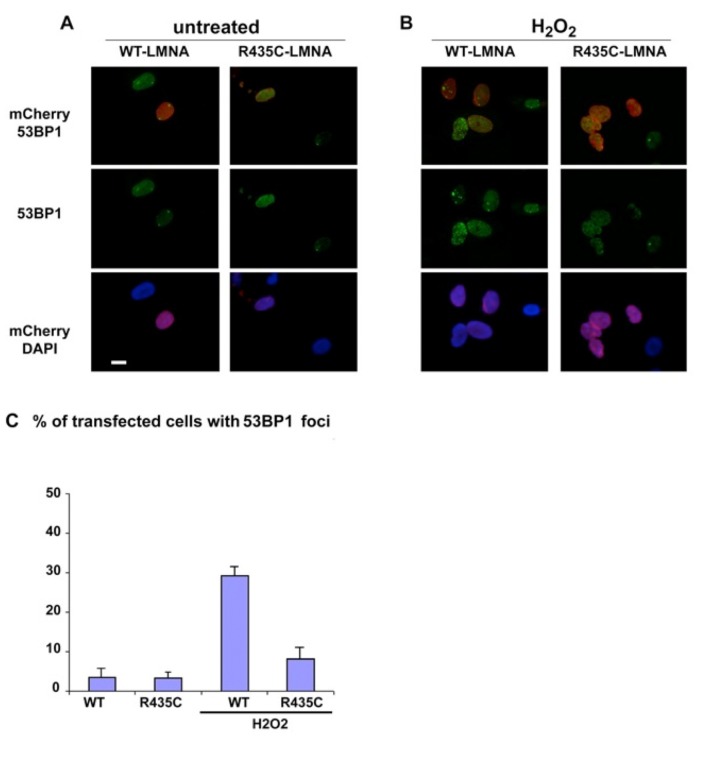
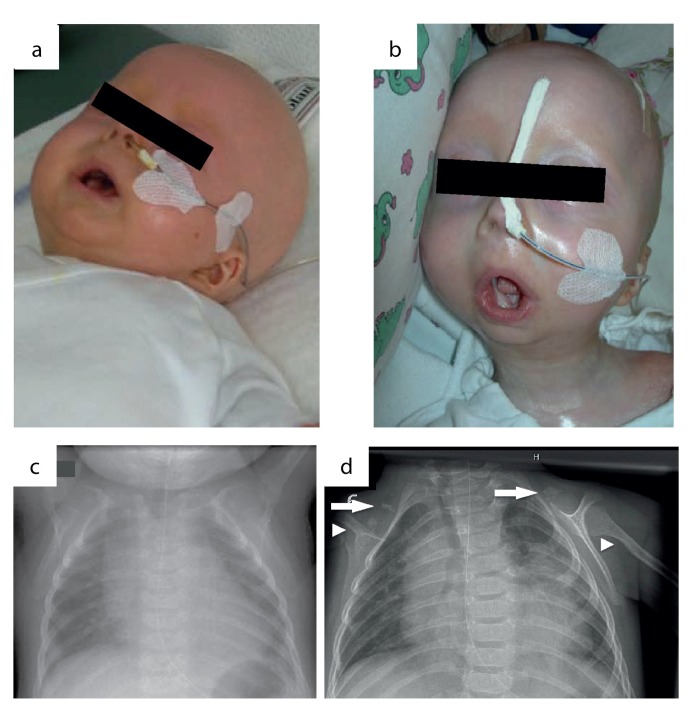
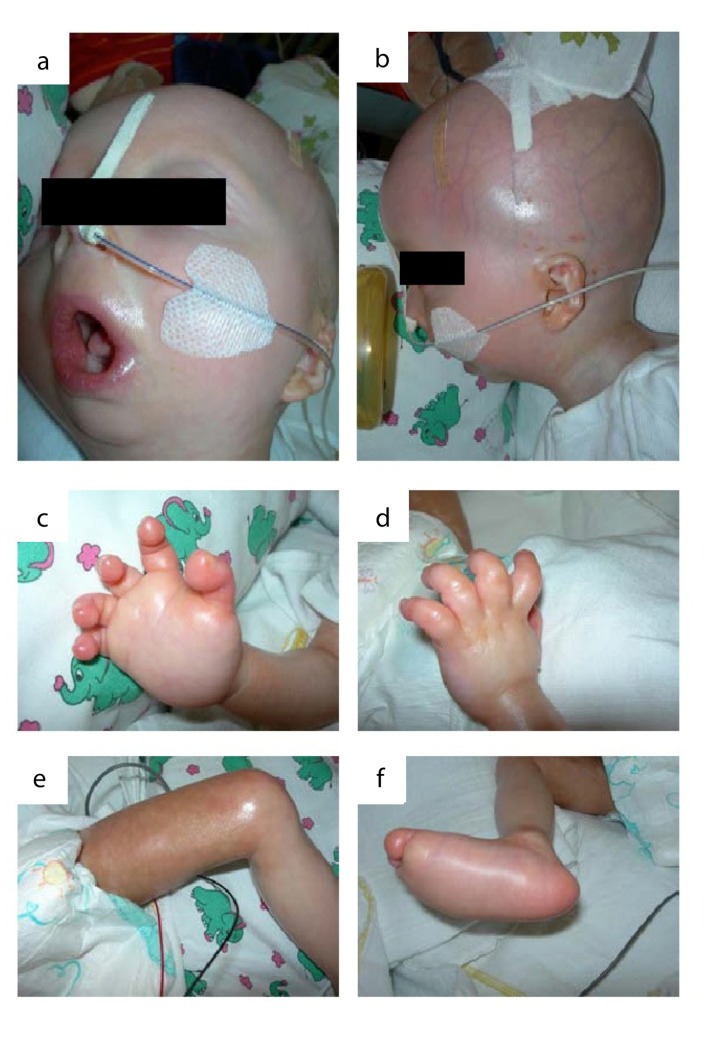
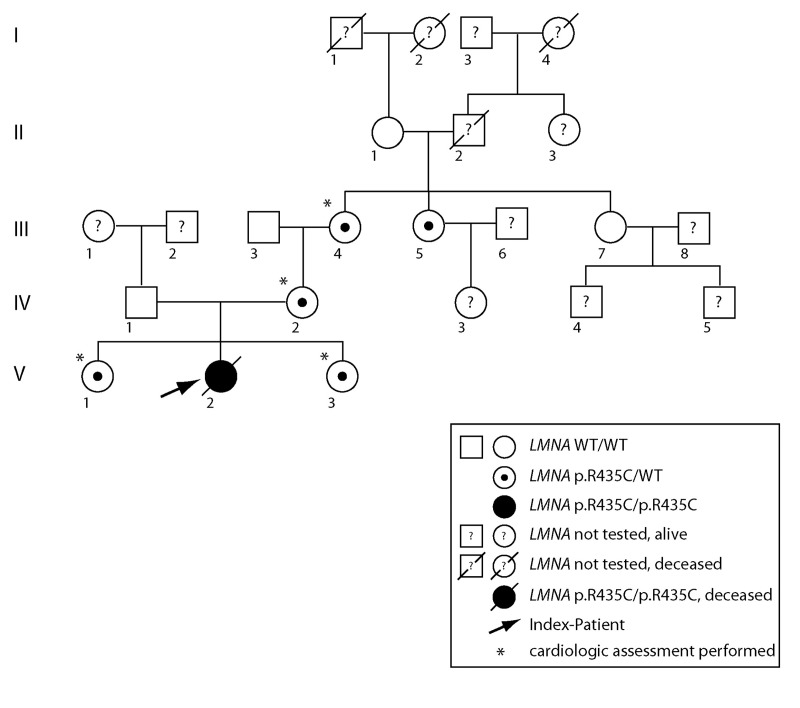
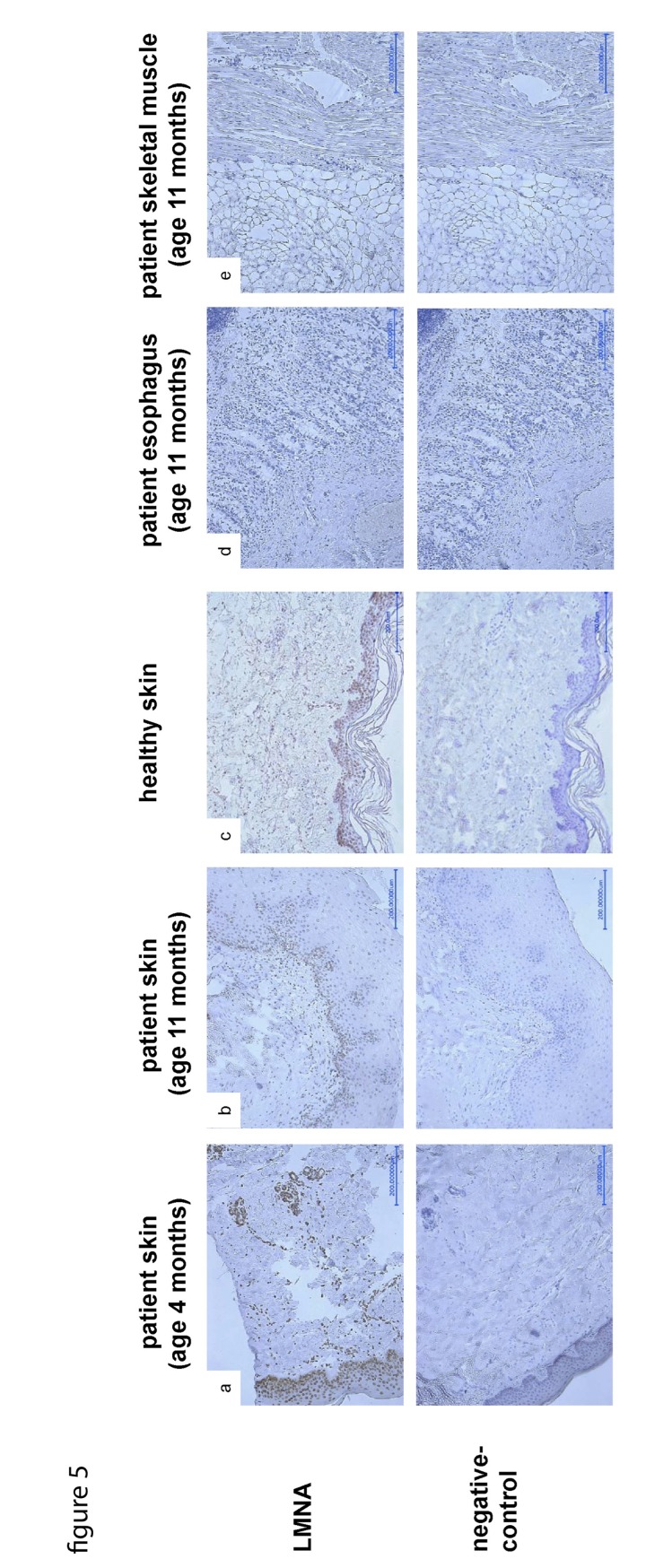
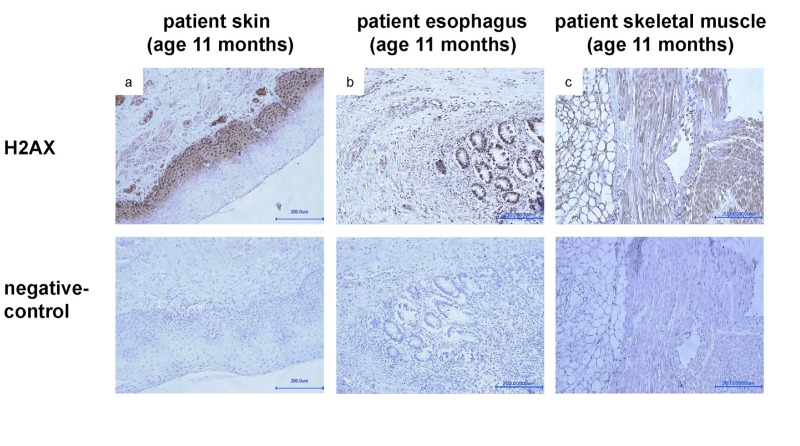
The clinical course of a female patient affected by a progeroid syndrome with Restrictive Dermopathy (RD)-like features was followed up. Besides missing hairiness, stagnating weight and growth, RD-like features including progressive skin swelling and solidification, acrocontractures, osteolysis and muscular hypotension were observed until the patient died at the age of 11 months. A homozygousLMNA mutation c.1303C>T (p.R435C) was found by Sanger sequencing. Haplotyping revealed a partial uniparental disomy of chromosome 1 (1q21.3 to 1q23.1) including the LMNA gene. In contrast to reported RD patients with LMNA mutations, LMNA p.R435C is not located at the cleavage site necessary for processing of prelamin A by ZMPSTE24 and leads to a distinct phenotype combining clinical features of Restrictive Dermopathy, Mandibuloacral Dysplasia and Hutchinson-Gilford Progeria. Functionally, LMNA p.R435C is associated with increasing DNA double strand breaks and decreased recruitment of P53 binding protein 1 (53BP1) to DNA-damage sites indicating delayed DNA repair. The follow-up of the complete clinical course in the patient combined with functional studies showed for the first time that a progressive loss of lamin A rather than abnormal accumulation of prelamin A species could be a pathophysiological mechanism in progeroid laminopathies, which leads to DNA repair deficiency accompanied by advancing tissue degeneration.
对一名患有具有限制性皮肤病(RD)样特征的早老症综合征女性患者的临床病程进行了随访。除了毛发缺失、体重和生长停滞外,还观察到了RD样特征,包括进行性皮肤肿胀和硬化、肢端挛缩、骨质溶解和肌肉张力减退,直至患者在11个月大时死亡。通过桑格测序发现了纯合的LMNA突变c.1303C>T(p.R435C)。单倍型分析显示1号染色体(1q21.3至1q23.1)包括LMNA基因存在部分单亲二体性。与报道的具有LMNA突变的RD患者不同,LMNA p.R435C并不位于ZMPSTE24处理前体核纤层蛋白A所需的切割位点,导致了一种独特的表型,结合了限制性皮肤病、下颌骨发育不全和哈钦森-吉尔福德早衰症的临床特征。在功能上,LMNA p.R435C与DNA双链断裂增加以及P53结合蛋白1(53BP1)向DNA损伤位点的募集减少有关,表明DNA修复延迟。对该患者完整临床病程的随访结合功能研究首次表明,核纤层蛋白A进行性丧失而非前体核纤层蛋白A物种的异常积累可能是早老症性核纤层蛋白病的病理生理机制,这导致DNA修复缺陷并伴有组织退行性变进展。