Meinke Peter, Hintze Stefan, Limmer Sarah, Schoser Benedikt
Friedrich-Baur-Institute at the Department of Neurology, University Hospital, Ludwig-Maximilians-University Munich, Munich, Germany.
Front Neurol. 2018 Jul 25;9:601. doi: 10.3389/fneur.2018.00601. eCollection 2018.
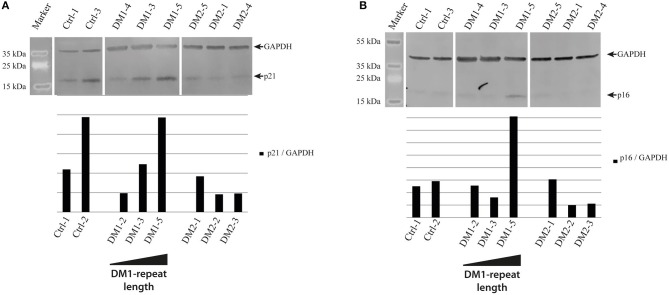
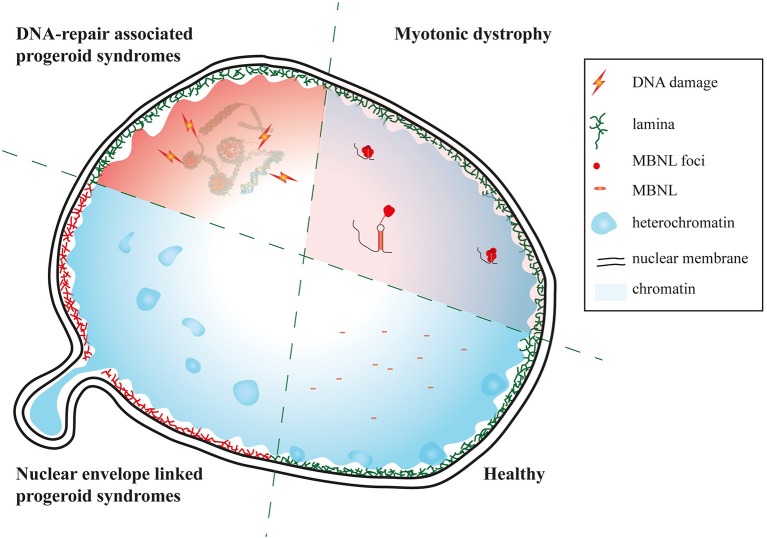
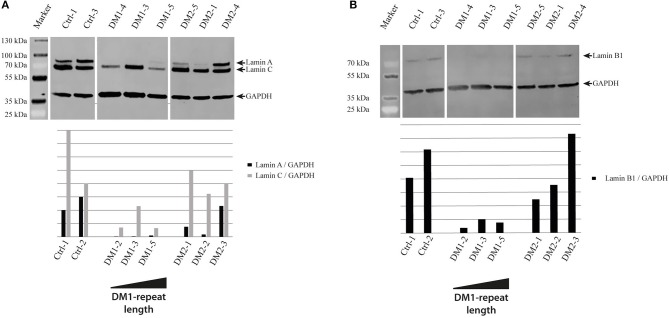
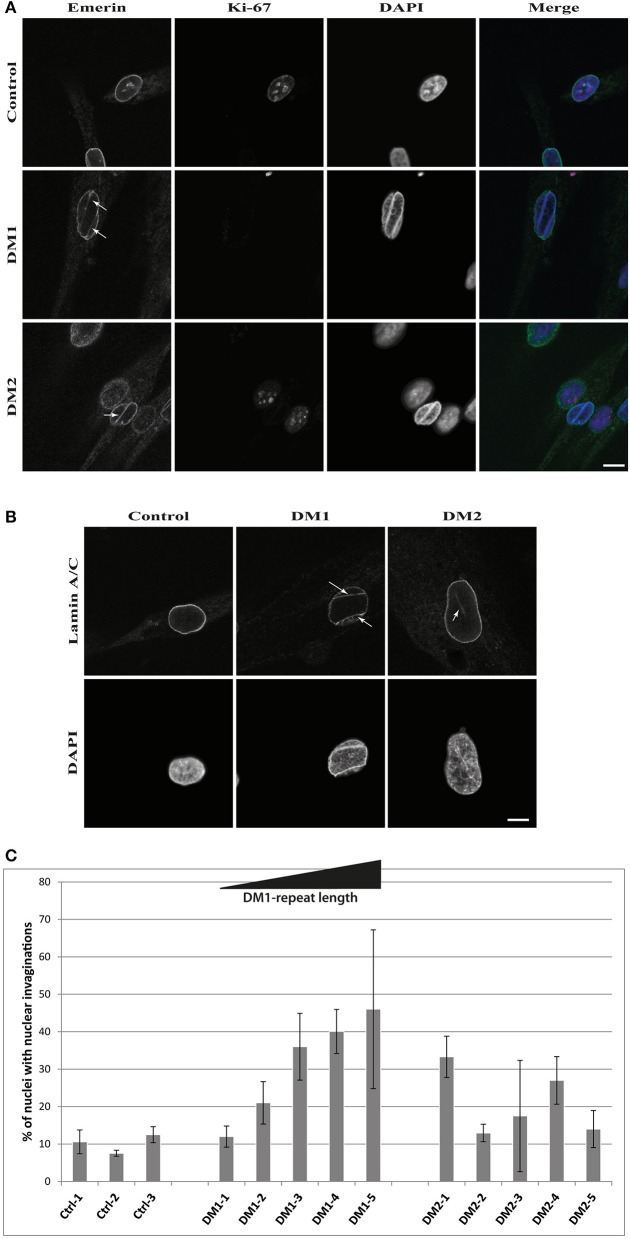
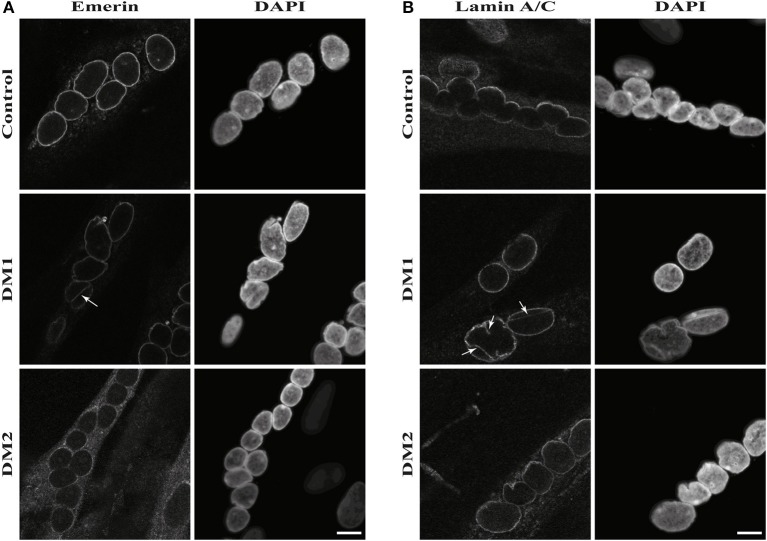
Myotonic dystrophies (DM) are slowly progressing multisystemic disorders caused by repeat expansions in the or genes. The multisystemic involvement in DM patients often reflects the appearance of accelerated aging. This is partly due to visible features such as cataracts, muscle weakness, and frontal baldness, but there are also less obvious features like cardiac arrhythmia, diabetes or hypogammaglobulinemia. These aging features suggest the hypothesis that DM could be a segmental progeroid disease. To identify the molecular cause of this characteristic appearance of accelerated aging we compare clinical features of DM to "typical" segmental progeroid disorders caused by mutations in DNA repair or nuclear envelope proteins. Furthermore, we characterize if this premature aging effect is also reflected on the cellular level in DM and investigate overlaps with "classical" progeroid disorders. To investigate the molecular similarities at the cellular level we use primary DM and control cell lines. This analysis reveals many similarities to progeroid syndromes linked to the nuclear envelope. Our comparison on both clinical and molecular levels argues for qualification of DM as a segmental progeroid disorder.
强直性肌营养不良症(DM)是由 或 基因中的重复序列扩增引起的缓慢进展的多系统疾病。DM患者的多系统受累往往反映出加速衰老的表现。这部分是由于白内障、肌肉无力和前额秃发等明显特征,但也有不太明显的特征,如心律失常、糖尿病或低丙种球蛋白血症。这些衰老特征提示了一个假说,即DM可能是一种节段性早老样疾病。为了确定这种加速衰老特征性表现的分子原因,我们将DM的临床特征与由DNA修复或核膜蛋白突变引起的“典型”节段性早老样疾病进行比较。此外,我们还确定这种早衰效应在DM的细胞水平上是否也有体现,并研究与“经典”早老样疾病的重叠情况。为了在细胞水平上研究分子相似性,我们使用原发性DM细胞系和对照细胞系。该分析揭示了与核膜相关的早老样综合征有许多相似之处。我们在临床和分子水平上的比较支持将DM定性为节段性早老样疾病。