Kilpatrick Alastair M, Ward Bruce, Aitken Stuart
School of Informatics, University of Edinburgh, Informatics Forum, 10 Crichton Street, EH8 9AB Edinburgh, Scotland.
Algorithms Mol Biol. 2013 Jun 27;8(1):16. doi: 10.1186/1748-7188-8-16.
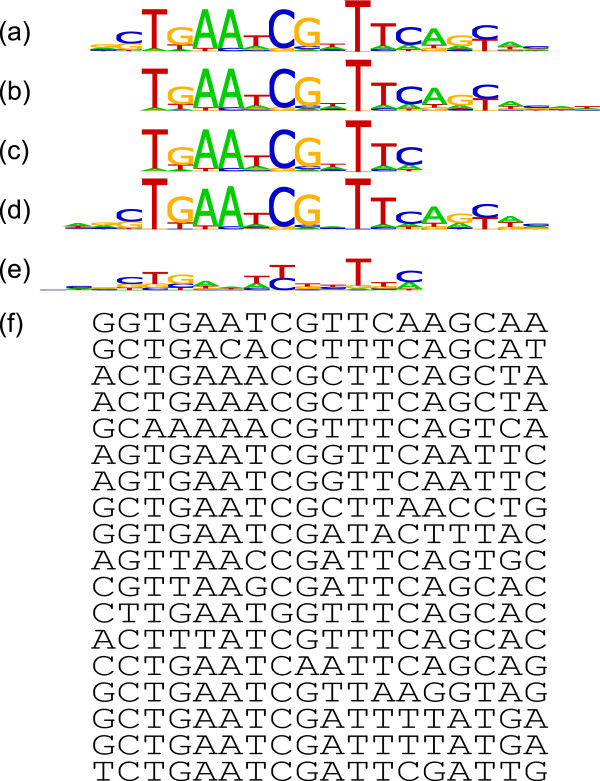
In transcription factor binding site discovery, the true width of the motif to be discovered is generally not known a priori. The ability to compute the most likely width of a motif is therefore a highly desirable property for motif discovery algorithms. However, this is a challenging computational problem as a result of changing model dimensionality at changing motif widths. The complexity of the problem is increased as the discovered model at the true motif width need not be the most statistically significant in a set of candidate motif models. Further, the core motif discovery algorithm used cannot guarantee to return the best possible result at each candidate width.
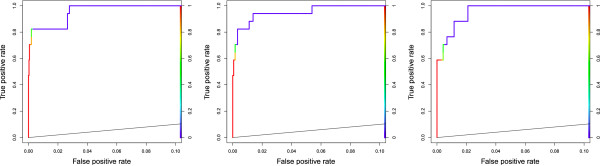
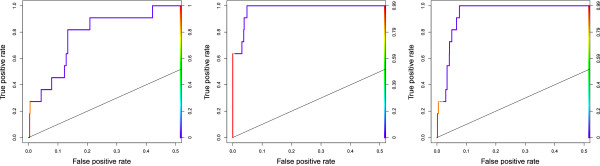
We present MCOIN, a novel heuristic for automatically determining transcription factor binding site motif width, based on motif containment and information content. Using realistic synthetic data and previously characterised prokaryotic data, we show that MCOIN outperforms the current most popular method (E-value of the resulting multiple alignment) as a predictor of motif width, based on mean absolute error. MCOIN is also shown to choose models which better match known sites at higher levels of motif conservation, based on ROC analysis.
We demonstrate the performance of MCOIN as part of a deterministic motif discovery algorithm and conclude that MCOIN outperforms current methods for determining motif width.
在转录因子结合位点发现中,待发现基序的真实宽度通常事先并不知晓。因此,对于基序发现算法而言,能够计算出最可能的基序宽度是一项非常理想的特性。然而,由于基序宽度变化时模型维度也会改变,这是一个具有挑战性的计算问题。该问题的复杂性还因在真实基序宽度下发现的模型在一组候选基序模型中不一定是统计意义最显著的而增加。此外,所使用的核心基序发现算法无法保证在每个候选宽度下都返回最佳可能结果。
我们提出了MCOIN,一种基于基序包含和信息含量自动确定转录因子结合位点基序宽度的新型启发式方法。使用逼真的合成数据和先前已表征的原核生物数据,基于平均绝对误差,我们表明MCOIN作为基序宽度预测器优于当前最流行的方法(所得多序列比对的E值)。基于ROC分析,MCOIN还表明在更高基序保守水平下能选择与已知位点更匹配的模型。
我们展示了MCOIN作为确定性基序发现算法一部分的性能,并得出结论:MCOIN在确定基序宽度方面优于当前方法。