Department of Genome Sciences, University of Washington, Seattle, Washington 98105, USA.
Genome Res. 2013 Sep;23(9):1373-82. doi: 10.1101/gr.158543.113. Epub 2013 Jul 3.
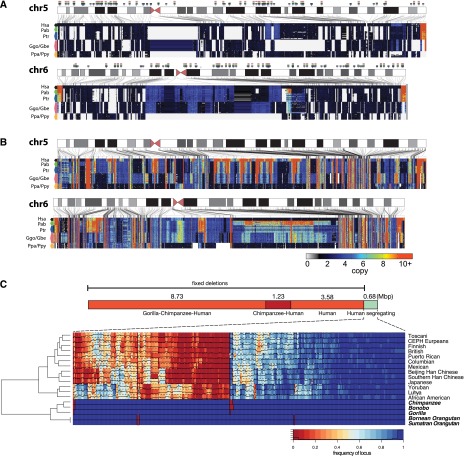
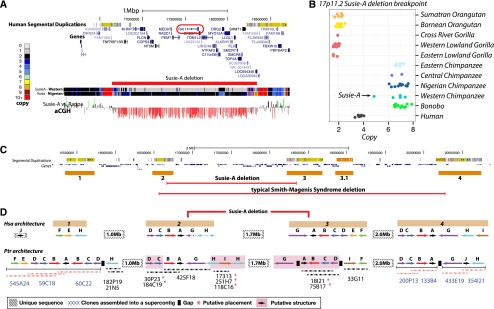
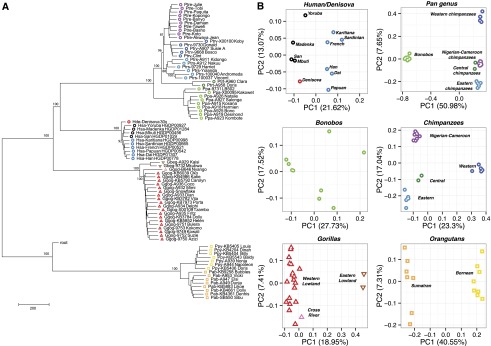
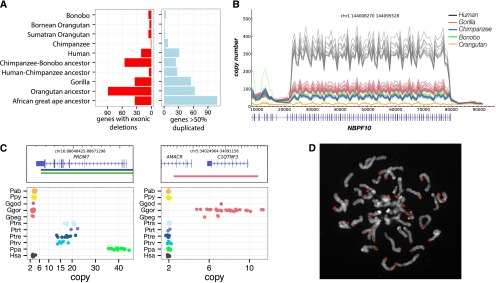
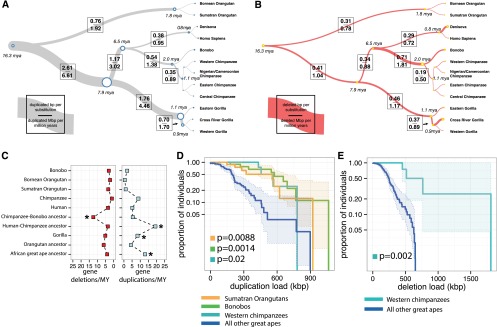
Copy number variation (CNV) contributes to disease and has restructured the genomes of great apes. The diversity and rate of this process, however, have not been extensively explored among great ape lineages. We analyzed 97 deeply sequenced great ape and human genomes and estimate 16% (469 Mb) of the hominid genome has been affected by recent CNV. We identify a comprehensive set of fixed gene deletions (n = 340) and duplications (n = 405) as well as >13.5 Mb of sequence that has been specifically lost on the human lineage. We compared the diversity and rates of copy number and single nucleotide variation across the hominid phylogeny. We find that CNV diversity partially correlates with single nucleotide diversity (r(2) = 0.5) and recapitulates the phylogeny of apes with few exceptions. Duplications significantly outpace deletions (2.8-fold). The load of segregating duplications remains significantly higher in bonobos, Western chimpanzees, and Sumatran orangutans-populations that have experienced recent genetic bottlenecks (P = 0.0014, 0.02, and 0.0088, respectively). The rate of fixed deletion has been more clocklike with the exception of the chimpanzee lineage, where we observe a twofold increase in the chimpanzee-bonobo ancestor (P = 4.79 × 10(-9)) and increased deletion load among Western chimpanzees (P = 0.002). The latter includes the first genomic disorder in a chimpanzee with features resembling Smith-Magenis syndrome mediated by a chimpanzee-specific increase in segmental duplication complexity. We hypothesize that demographic effects, such as bottlenecks, have contributed to larger and more gene-rich segments being deleted in the chimpanzee lineage and that this effect, more generally, may account for episodic bursts in CNV during hominid evolution.
拷贝数变异 (CNV) 导致疾病,并重构了大猿类的基因组。然而,在大猿类谱系中,这一过程的多样性和速率尚未得到广泛探索。我们分析了 97 个深度测序的大猿和人类基因组,并估计人类基因组中有 16%(469Mb)受到了最近 CNV 的影响。我们确定了一套全面的固定基因缺失(n=340)和重复(n=405),以及人类谱系中特异性丢失的超过 13.5Mb 的序列。我们比较了同源人科进化枝中拷贝数和单核苷酸变异的多样性和速率。我们发现,CNV 多样性部分与单核苷酸多样性相关(r(2)=0.5),并重现了除少数例外之外的猿类进化史。重复明显快于缺失(2.8 倍)。在经历了最近遗传瓶颈的倭黑猩猩、西部黑猩猩和苏门答腊猩猩群体中,分离重复的负荷仍然明显更高(P=0.0014、0.02 和 0.0088)。除了黑猩猩谱系外,固定缺失的速率一直更像时钟,在黑猩猩-倭黑猩猩祖先中观察到两倍的增加(P=4.79×10(-9)),并且西部黑猩猩中的缺失负荷增加(P=0.002)。后者包括了第一个在黑猩猩中出现的基因组疾病,其特征类似于由黑猩猩特异性的片段重复复杂性增加介导的 Smith-Magenis 综合征。我们假设,人口统计学效应,如瓶颈,导致了黑猩猩谱系中更大和更多基因丰富的片段被删除,并且这种效应更普遍地可能解释了人类进化过程中 CNV 的突发。