Department of Biostatistics and Epidemiology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, United States of America.
PLoS One. 2013 Jun 24;8(6):e66883. doi: 10.1371/journal.pone.0066883. Print 2013.
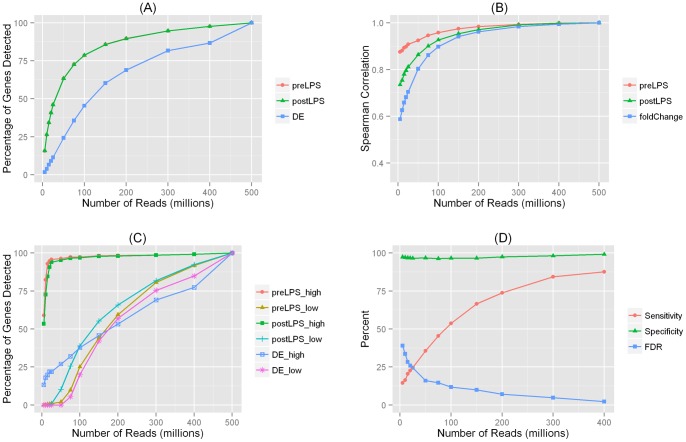
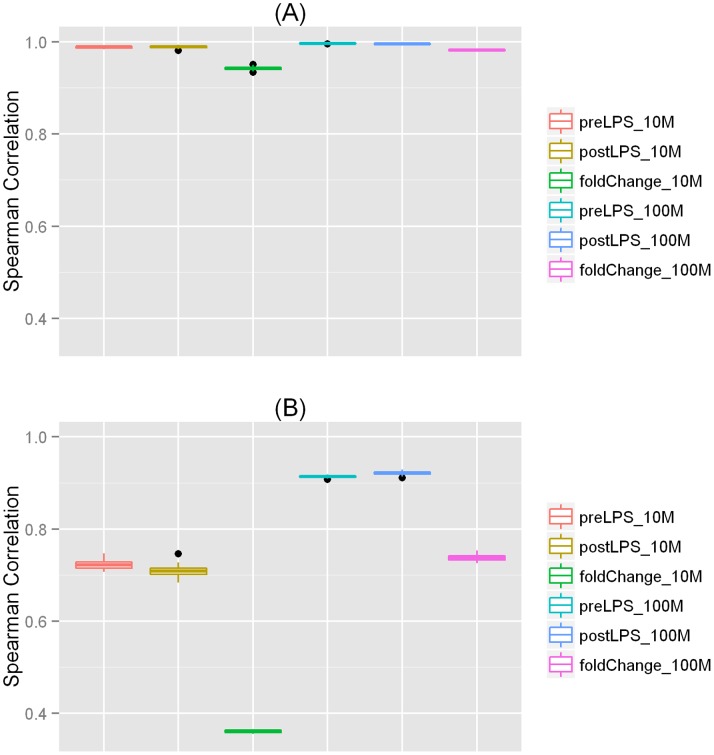
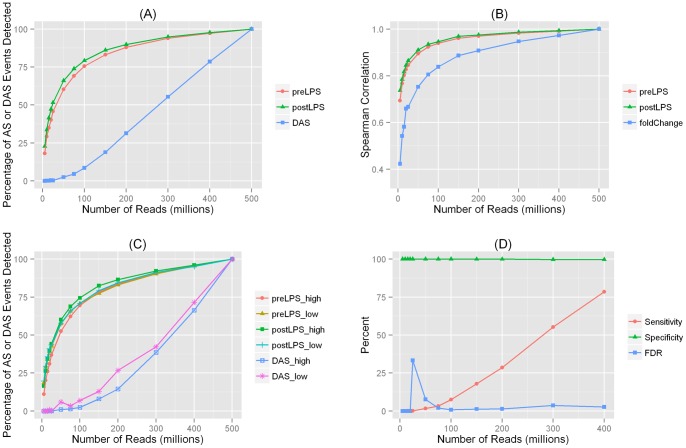
Recent advances in RNA sequencing (RNA-Seq) have enabled the discovery of novel transcriptomic variations that are not possible with traditional microarray-based methods. Tissue and cell specific transcriptome changes during pathophysiological stress in disease cases versus controls and in response to therapies are of particular interest to investigators studying cardiometabolic diseases. Thus, knowledge on the relationships between sequencing depth and detection of transcriptomic variation is needed for designing RNA-Seq experiments and for interpreting results of analyses. Using deeply sequenced Illumina HiSeq 2000 101 bp paired-end RNA-Seq data derived from adipose of a healthy individual before and after systemic administration of endotoxin (LPS), we investigated the sequencing depths needed for studies of gene expression and alternative splicing (AS). In order to detect expressed genes and AS events, we found that ∼100 to 150 million (M) filtered reads were needed. However, the requirement on sequencing depth for the detection of LPS modulated differential expression (DE) and differential alternative splicing (DAS) was much higher. To detect 80% of events, ∼300 M filtered reads were needed for DE analysis whereas at least 400 M filtered reads were necessary for detecting DAS. Although the majority of expressed genes and AS events can be detected with modest sequencing depths (∼100 M filtered reads), the estimated gene expression levels and exon/intron inclusion levels were less accurate. We report the first study that evaluates the relationship between RNA-Seq depth and the ability to detect DE and DAS in human adipose. Our results suggest that a much higher sequencing depth is needed to reliably identify DAS events than for DE genes.
近年来,RNA 测序(RNA-Seq)的进展使得发现新型转录组变异成为可能,而这些变异是传统基于微阵列的方法无法实现的。在疾病病例与对照以及对治疗的反应中,组织和细胞特定的转录组在病理生理应激下的变化,是研究心脏代谢疾病的研究人员特别感兴趣的。因此,在设计 RNA-Seq 实验和解释分析结果时,需要了解测序深度与转录组变异检测之间的关系。我们使用深度测序的 Illumina HiSeq 2000 101 个碱基对的配对末端 RNA-Seq 数据,该数据来自于健康个体在全身给予内毒素(LPS)前后的脂肪组织,研究了用于基因表达和选择性剪接(AS)研究的测序深度。为了检测表达基因和 AS 事件,我们发现需要约 1 亿到 1.5 亿个过滤后的读取。然而,对于检测 LPS 调节的差异表达(DE)和差异选择性剪接(DAS),测序深度的要求要高得多。为了检测 80%的事件,DE 分析需要约 3 亿个过滤后的读取,而检测 DAS 则至少需要 4 亿个过滤后的读取。虽然大多数表达基因和 AS 事件可以用适度的测序深度(约 1 亿个过滤后的读取)检测到,但估计的基因表达水平和外显子/内含子包含水平不太准确。我们报告了第一项评估 RNA-Seq 深度与在人类脂肪中检测 DE 和 DAS 能力之间关系的研究。我们的结果表明,可靠地识别 DAS 事件所需的测序深度比 DE 基因高得多。