Department of Crop Sciences, University of Illinois, Urbana, IL 61801, USA.
BMC Genomics. 2013 Jul 16;14:477. doi: 10.1186/1471-2164-14-477.
Two plant-specific transcription factors, NAC and YABBY, are involved in important plant developmental processes. However their molecular mechanisms, especially DNA binding sites and co-regulated genes, are largely unknown during soybean seedling development.
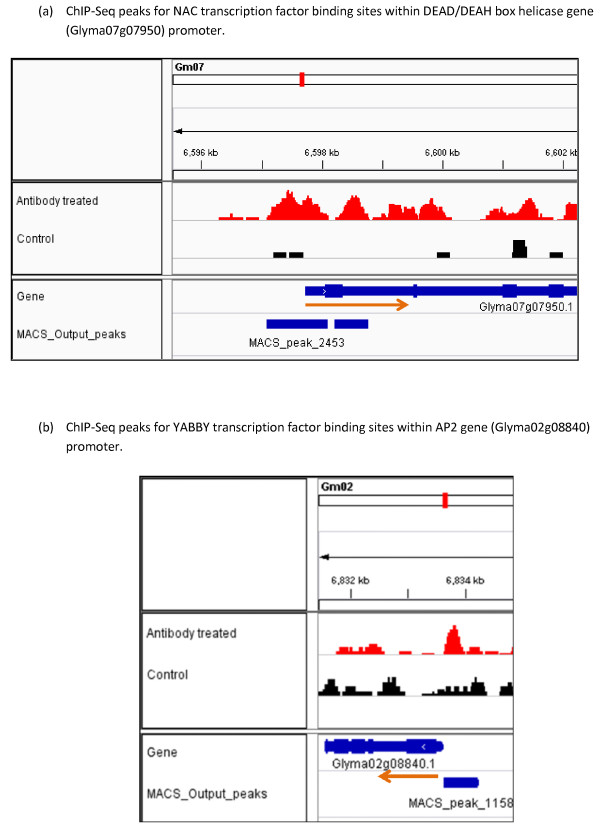
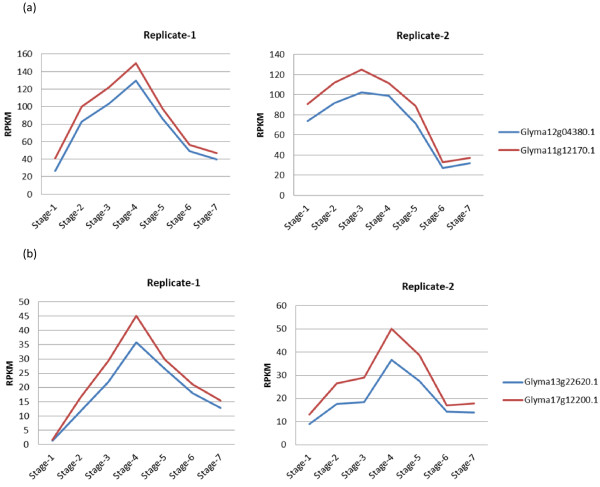
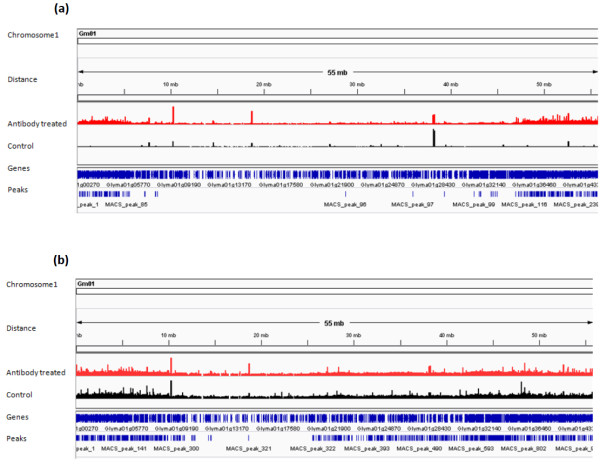
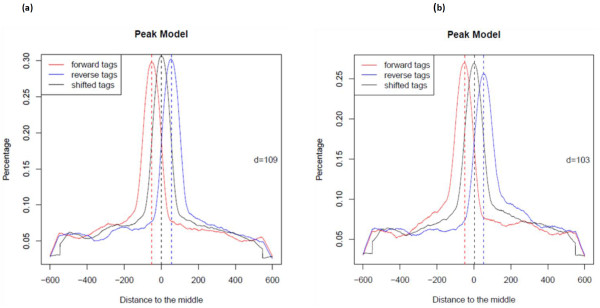
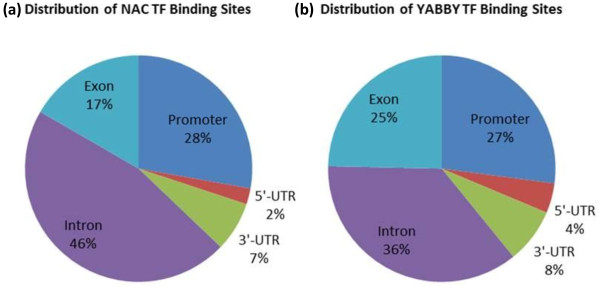
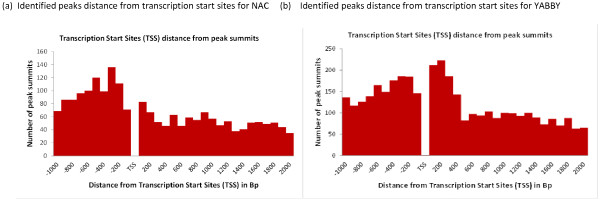
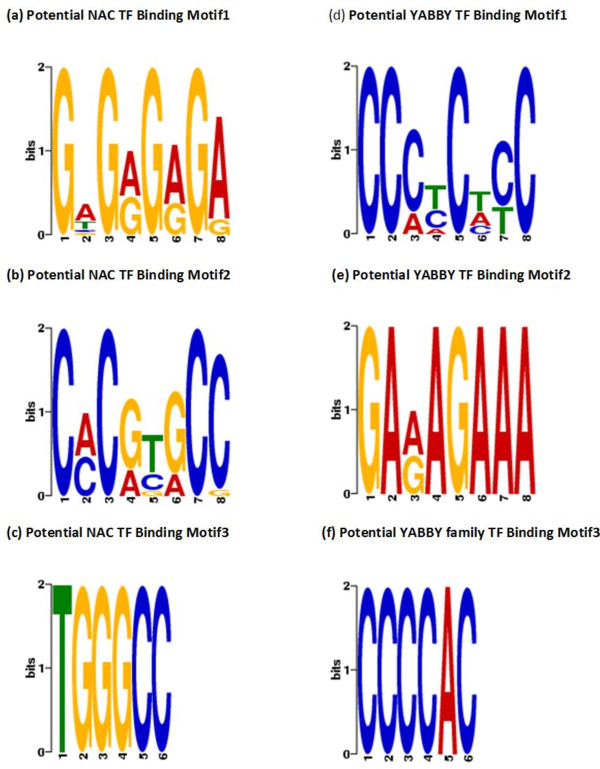
In order to identify genome-wide binding sites of specific members of the NAC and YABBY transcription factors and co-regulated genes, we performed Chromatin Immunoprecipitation Sequencing (ChIP-Seq) and RNA Sequencing (RNA-Seq) using cotyledons from soybean seedling developmental stages. Our RNA-Seq data revealed that these particular NAC and YABBY transcription factors showed a clear pattern in their expression during soybean seedling development. The highest level of their expression was found in seedling developmental stage 4 when cotyledons undergo a physiological transition from non-photosynthetic storage tissue to a metabolically active photosynthetic tissue. Our ChIP-Seq data identified 72 genes potentially regulated by the NAC and 96 genes by the YABBY transcription factors examined. Our RNA-Seq data revealed highly differentially expressed candidate genes regulated by the NAC transcription factor include lipoxygense, pectin methyl esterase inhibitor, DEAD/DEAH box helicase and homeobox associated proteins. YABBY-regulated genes include AP2 transcription factor, fatty acid desaturase and WRKY transcription factor. Additionally, we have identified DNA binding motifs for the NAC and YABBY transcription factors.
Genome-wide determination of binding sites for NAC and YABBY transcription factors and identification of candidate genes regulated by these transcription factors will advance the understanding of complex gene regulatory networks during soybean seedling development. Our data imply that there is transcriptional reprogramming during the functional transition of cotyledons from non-photosynthetic storage tissue to metabolically active photosynthetic tissue.
两个植物特异性转录因子 NAC 和 YABBY 参与了重要的植物发育过程。然而,它们的分子机制,特别是 DNA 结合位点和共同调控的基因,在大豆幼苗发育过程中很大程度上是未知的。
为了鉴定 NAC 和 YABBY 转录因子特定成员的全基因组结合位点和共同调控的基因,我们使用 cotyledons 来自大豆幼苗发育阶段进行了染色质免疫沉淀测序(ChIP-Seq)和 RNA 测序(RNA-Seq)。我们的 RNA-Seq 数据表明,这些特定的 NAC 和 YABBY 转录因子在大豆幼苗发育过程中的表达呈现出明显的模式。当子叶经历从非光合贮藏组织到代谢活跃的光合组织的生理转变时,它们的表达水平在幼苗发育阶段 4 达到最高。我们的 ChIP-Seq 数据鉴定了 72 个可能受 NAC 调控的基因和 96 个受 YABBY 转录因子调控的基因。我们的 RNA-Seq 数据揭示了受 NAC 转录因子调控的高度差异表达的候选基因,包括 lipoxygense、pectin methyl esterase inhibitor、DEAD/DEAH box helicase 和 homeobox associated proteins。YABBY 调控的基因包括 AP2 转录因子、脂肪酸去饱和酶和 WRKY 转录因子。此外,我们还确定了 NAC 和 YABBY 转录因子的 DNA 结合基序。
NAC 和 YABBY 转录因子结合位点的全基因组测定以及这些转录因子调控的候选基因的鉴定,将推进对大豆幼苗发育过程中复杂基因调控网络的理解。我们的数据表明,在子叶从非光合贮藏组织到代谢活跃的光合组织的功能转变过程中存在转录重编程。