Department of Biomedical Engineering, Yale University, New Haven, Connecticut, USA.
PLoS Comput Biol. 2013;9(7):e1003135. doi: 10.1371/journal.pcbi.1003135. Epub 2013 Jul 11.
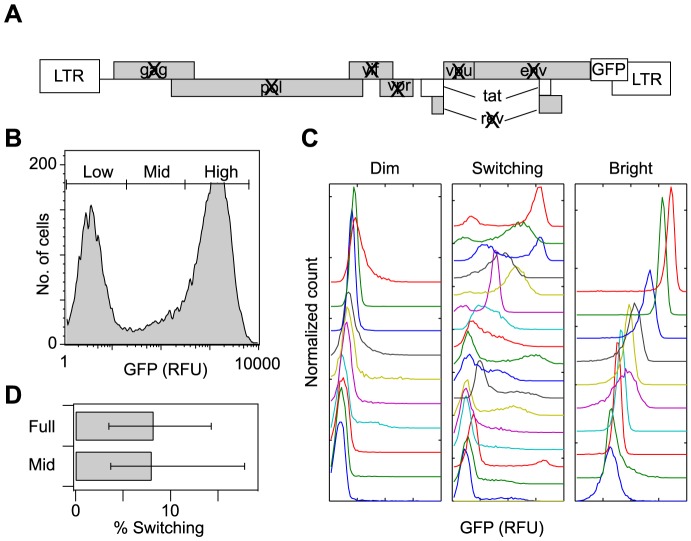
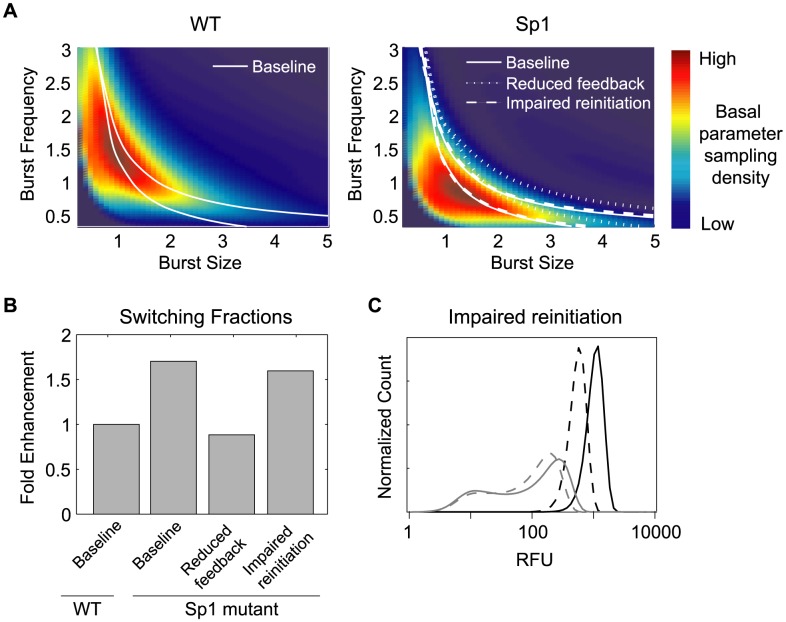
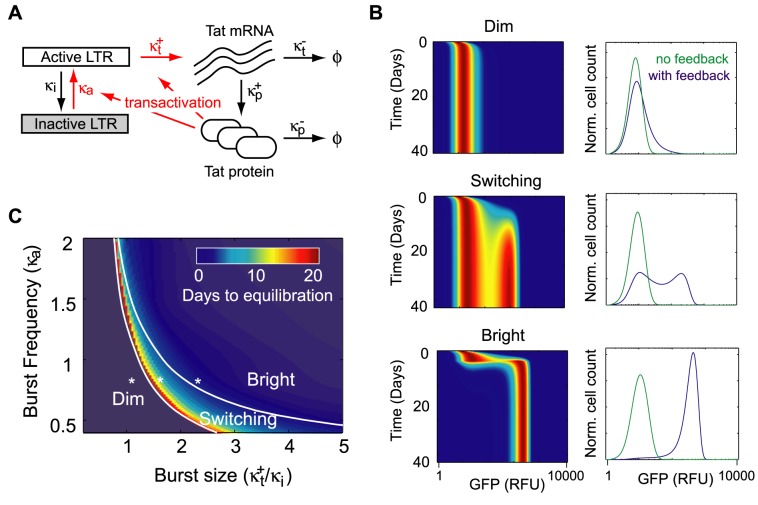
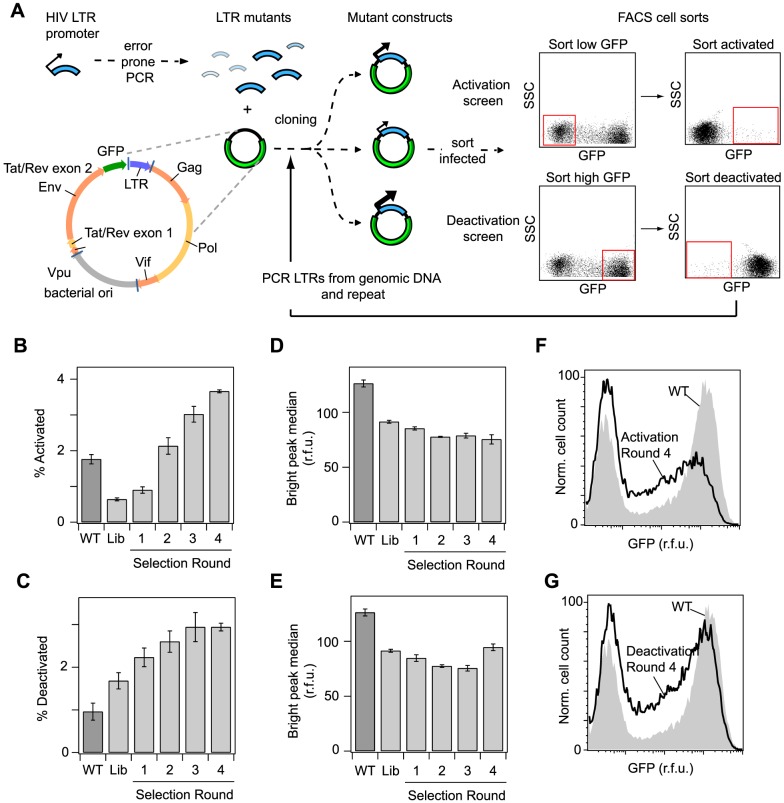
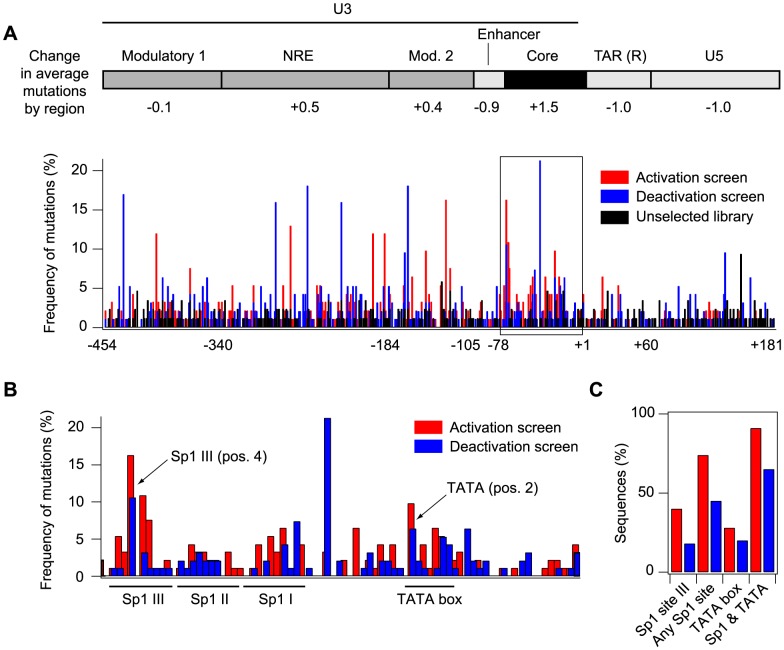
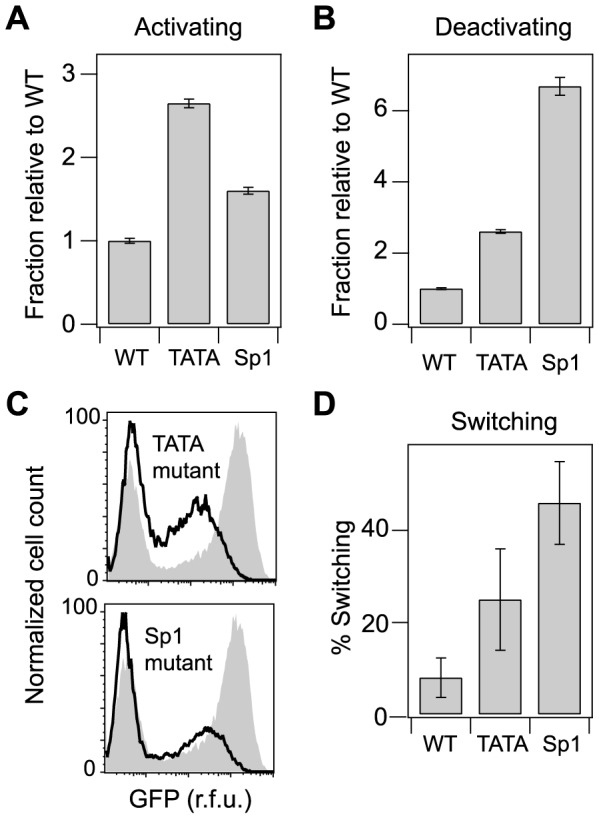
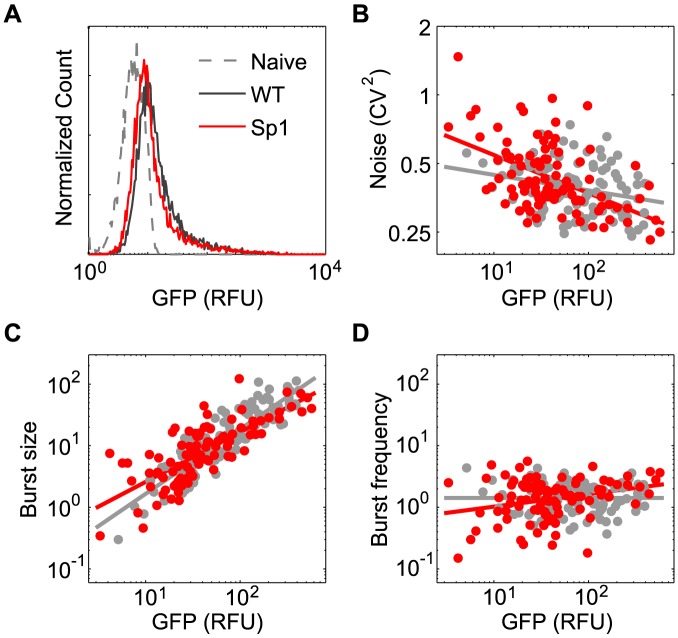
The sequence of a promoter within a genome does not uniquely determine gene expression levels and their variability; rather, promoter sequence can additionally interact with its location in the genome, or genomic context, to shape eukaryotic gene expression. Retroviruses, such as human immunodeficiency virus-1 (HIV), integrate their genomes into those of their host and thereby provide a biomedically-relevant model system to quantitatively explore the relationship between promoter sequence, genomic context, and noise-driven variability on viral gene expression. Using an in vitro model of the HIV Tat-mediated positive-feedback loop, we previously demonstrated that fluctuations in viral Tat-transactivating protein levels generate integration-site-dependent, stochastically-driven phenotypes, in which infected cells randomly 'switch' between high and low expressing states in a manner that may be related to viral latency. Here we extended this model and designed a forward genetic screen to systematically identify genetic elements in the HIV LTR promoter that modulate the fraction of genomic integrations that specify 'Switching' phenotypes. Our screen identified mutations in core promoter regions, including Sp1 and TATA transcription factor binding sites, which increased the Switching fraction several fold. By integrating single-cell experiments with computational modeling, we further investigated the mechanism of Switching-fraction enhancement for a selected Sp1 mutation. Our experimental observations demonstrated that the Sp1 mutation both impaired Tat-transactivated expression and also altered basal expression in the absence of Tat. Computational analysis demonstrated that the observed change in basal expression could contribute significantly to the observed increase in viral integrations that specify a Switching phenotype, provided that the selected mutation affected Tat-mediated noise amplification differentially across genomic contexts. Our study thus demonstrates a methodology to identify and characterize promoter elements that affect the distribution of stochastic phenotypes over genomic contexts, and advances our understanding of how promoter mutations may control the frequency of latent HIV infection.
基因组中启动子的序列并不能唯一决定基因表达水平及其可变性;相反,启动子序列还可以与其在基因组中的位置(或基因组背景)相互作用,从而塑造真核基因表达。逆转录病毒,如人类免疫缺陷病毒 1(HIV),将其基因组整合到宿主的基因组中,从而为定量探索启动子序列、基因组背景和噪声驱动的病毒基因表达变异性之间的关系提供了一个有生物医学意义的模型系统。我们之前使用 HIV Tat 介导的正反馈环的体外模型证明,病毒 Tat 转录激活蛋白水平的波动会产生整合位点依赖性、随机驱动的表型,其中受感染的细胞以一种可能与病毒潜伏相关的方式随机“切换”到高表达和低表达状态。在这里,我们扩展了这个模型,并设计了一个正向遗传筛选,以系统地识别 HIV LTR 启动子中的遗传元件,这些元件可以调节指定“切换”表型的基因组整合的分数。我们的筛选确定了核心启动子区域中的突变,包括 Sp1 和 TATA 转录因子结合位点,这些突变将“切换”分数提高了几倍。通过将单细胞实验与计算模型相结合,我们进一步研究了选定 Sp1 突变的切换分数增强的机制。我们的实验观察表明,Sp1 突变既损害了 Tat 转录激活的表达,也改变了没有 Tat 的情况下的基础表达。计算分析表明,在不同基因组背景下,Tat 介导的噪声放大对观察到的基础表达变化的影响可能会显著影响指定切换表型的病毒整合数量,假设所选突变在不同基因组背景下对 Tat 介导的噪声放大有不同的影响。因此,我们的研究表明了一种识别和表征影响随机表型在基因组背景下分布的启动子元件的方法,并加深了我们对启动子突变如何控制潜伏性 HIV 感染频率的理解。