Laboratory for Molecular and Cell Biology, Paediatric Neurology Unit and Laboratories, Meyer Children's Hospital, Viale Pieraccini n, 24, Florence 50139, Italy.
Orphanet J Rare Dis. 2013 Aug 2;8:114. doi: 10.1186/1750-1172-8-114.
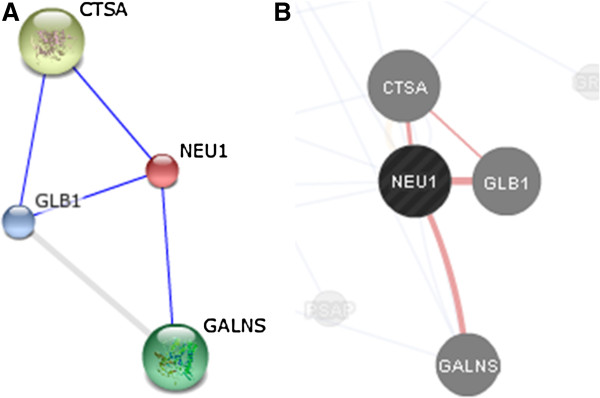
Mutations in the CTSA gene, that encodes the protective protein/cathepsin A or PPCA, lead to the secondary deficiency of β-galactosidase (GLB1) and neuraminidase 1 (NEU1), causing the lysosomal storage disorder galactosialidosis (GS). Few clinical cases of GS have been reported in the literature, the majority of them belonging to the juvenile/adult group of patients.
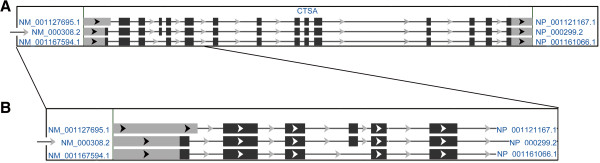
The correct nomenclature of mutations for this gene is discussed through the analysis of the three PPCA/CTSA isoforms available in the GenBank database. Phenotype-genotype correlation has been assessed by computational analysis and review of previously reported single amino acid substitutions.
We report the clinical and mutational analyses of four cases with the rare infantile form of GS. We identified three novel nucleotide changes, two of them resulting in the missense mutations, c.347A>G (p.His116Arg), c.775T>C (p.Cys259Arg), and the third, c.1216C>T, resulting in the p.Gln406* stop codon, a type of mutation identified for the first time in GS. An Italian founder effect of the c.114delG mutation can be suggested according to the origin of the only three patients carrying this mutation reported here and in the literature.
In early reports mutations nomenclature was selected according to all CTSA isoforms (three different isoforms), thus generating a lot of confusion. In order to assist physicians in the interpretation of detected mutations, we mark the correct nomenclature for CTSA mutations. The complexity of pathology caused by the multifunctions of CTSA, and the very low numbers of mutations (only 23 overall) in relation to the length of the CTSA gene are discussed.In addition, the in silico functional predictions of all reported missense mutations allowed us to closely predict the early infantile, late infantile and juvenile phenotypes, also disclosing different degrees of severity in the juvenile phenotype.
编码保护性蛋白/组织蛋白酶 A 或 PPCA 的 CTSA 基因突变导致β-半乳糖苷酶(GLB1)和神经氨酸酶 1(NEU1)的继发缺乏,从而导致溶酶体贮积病半乳糖脑苷脂病(GS)。文献中报道了少数 GS 临床病例,其中大多数属于青少年/成年患者。
通过分析 GenBank 数据库中可用的三种 PPCA/CTSA 同工型,讨论了该基因突变的正确命名。通过计算分析和回顾以前报道的单个氨基酸取代,评估了表型-基因型相关性。
我们报告了 4 例罕见婴儿型 GS 的临床和突变分析。我们确定了三个新的核苷酸变化,其中两个导致错义突变,c.347A>G(p.His116Arg),c.775T>C(p.Cys259Arg),第三个 c.1216C>T 导致 p.Gln406* 终止密码子,这是在 GS 中首次发现的一种突变类型。根据这里和文献中报道的仅携带这种突变的三个患者的起源,可以推测 c.114delG 突变存在意大利的创始人效应。
在早期报告中,突变命名是根据所有 CTSA 同工型(三种不同同工型)选择的,因此产生了很多混淆。为了帮助医生解释检测到的突变,我们标记了 CTSA 突变的正确命名。CTSA 多功能引起的病理学的复杂性,以及与 CTSA 基因长度相比非常少的突变(总共只有 23 个),都进行了讨论。此外,所有报道的错义突变的计算机功能预测使我们能够密切预测早发性婴儿型、晚发性婴儿型和青少年型表型,同时也揭示了青少年型表型的不同严重程度。