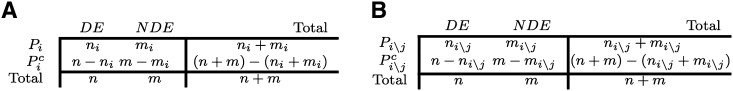Computer Science Department, Wayne State University, Detroit, Michigan 48084, USA;
Genome Res. 2013 Nov;23(11):1885-93. doi: 10.1101/gr.153551.112. Epub 2013 Aug 9.
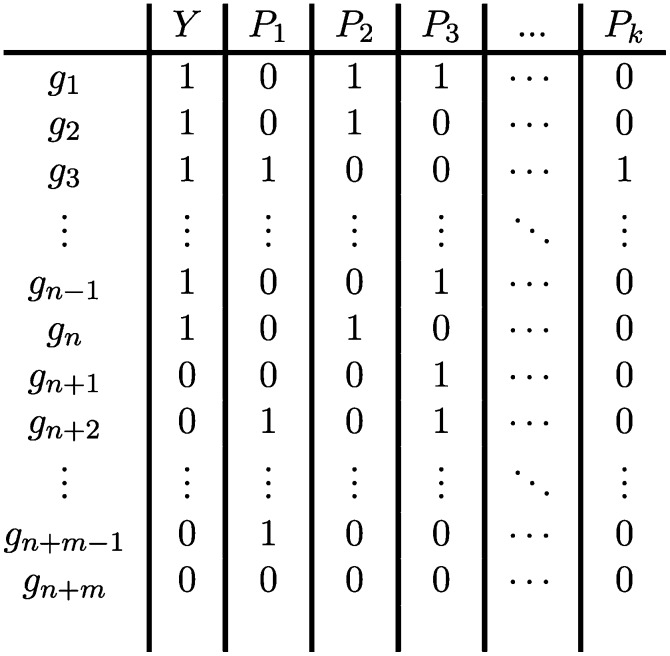
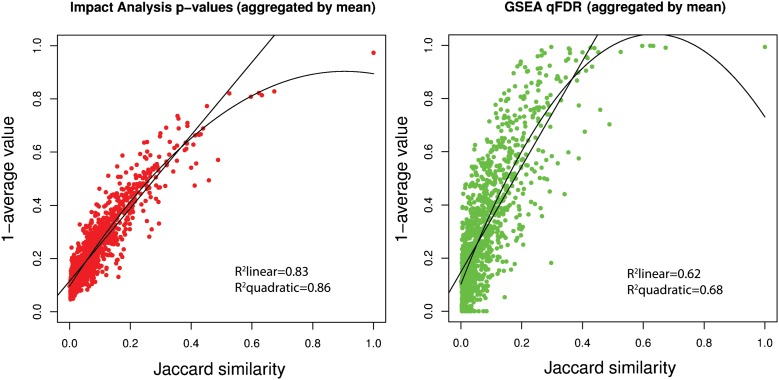
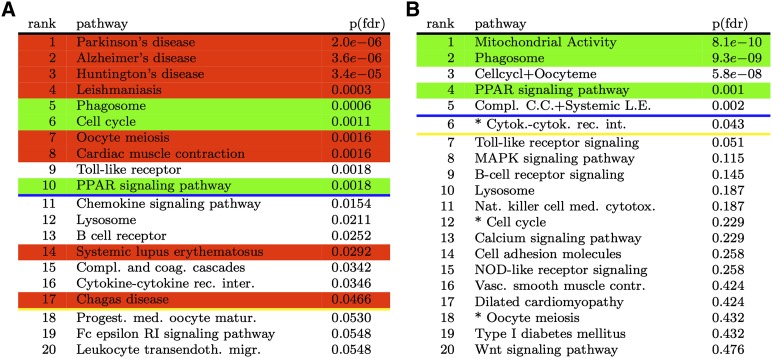
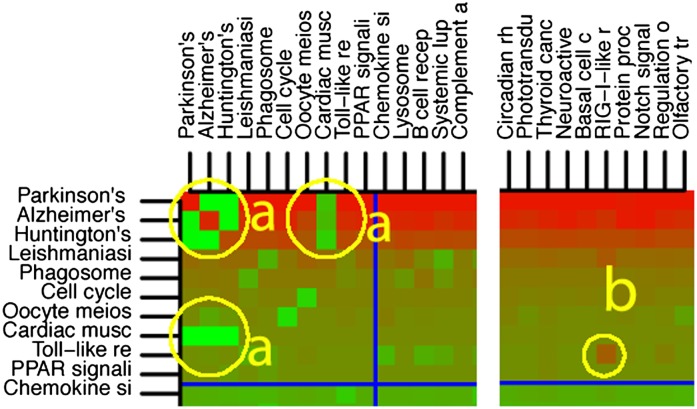
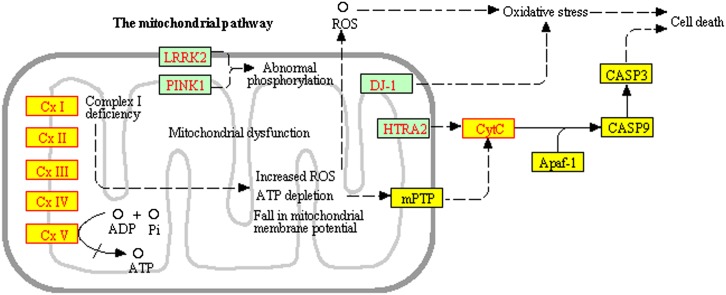
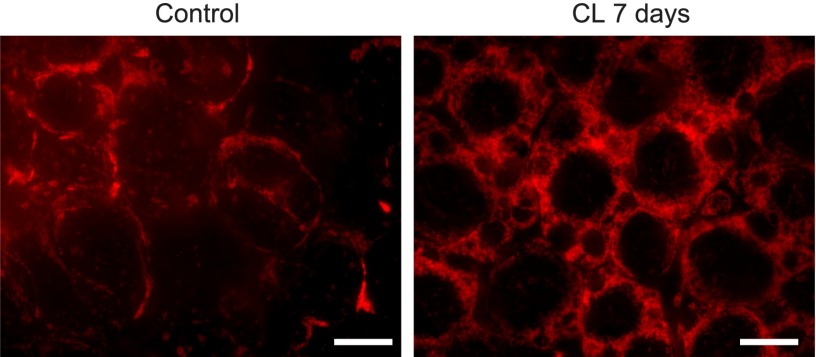
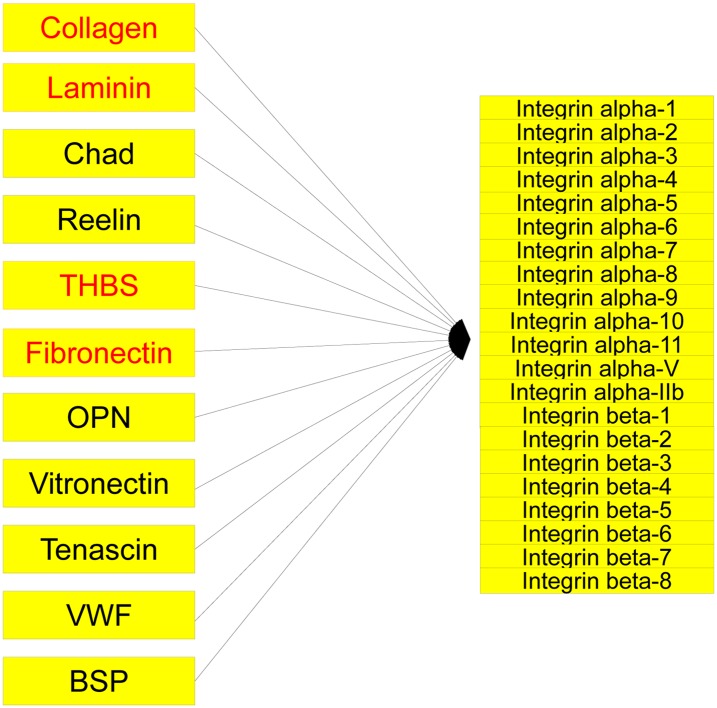
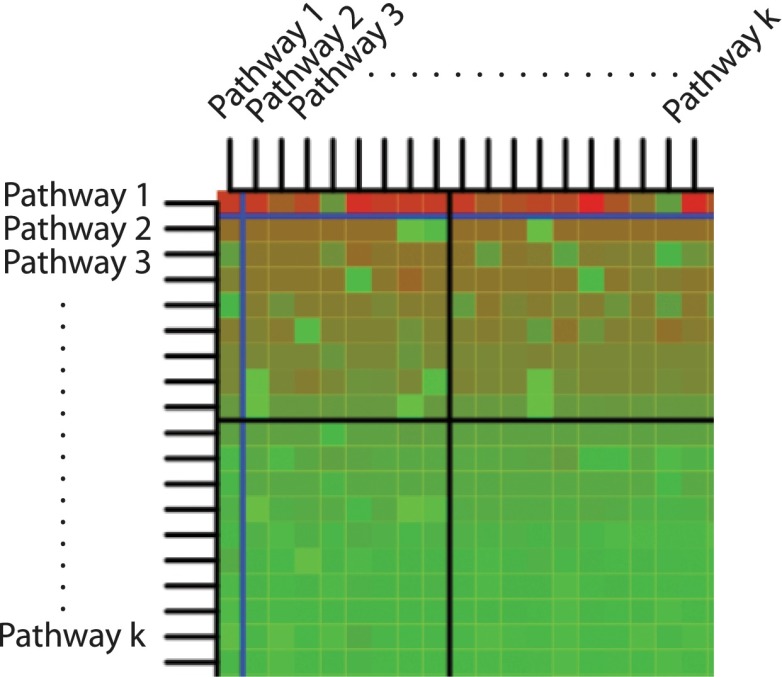
Identifying the pathways that are significantly impacted in a given condition is a crucial step in understanding the underlying biological phenomena. All approaches currently available for this purpose calculate a P-value that aims to quantify the significance of the involvement of each pathway in the given phenotype. These P-values were previously thought to be independent. Here we show that this is not the case, and that many pathways can considerably affect each other's P-values through a "crosstalk" phenomenon. Although it is intuitive that various pathways could influence each other, the presence and extent of this phenomenon have not been rigorously studied and, most importantly, there is no currently available technique able to quantify the amount of such crosstalk. Here, we show that all three major categories of pathway analysis methods (enrichment analysis, functional class scoring, and topology-based methods) are severely influenced by crosstalk phenomena. Using real pathways and data, we show that in some cases pathways with significant P-values are not biologically meaningful, and that some biologically meaningful pathways with nonsignificant P-values become statistically significant when the crosstalk effects of other pathways are removed. We describe a technique able to detect, quantify, and correct crosstalk effects, as well as identify independent functional modules. We assessed this novel approach on data from four experiments involving three phenotypes and two species. This method is expected to allow a better understanding of individual experiment results, as well as a more refined definition of the existing signaling pathways for specific phenotypes.
确定给定条件下受显著影响的途径是理解潜在生物学现象的关键步骤。目前所有用于此目的的方法都计算了一个 P 值,旨在量化每条途径在给定表型中的参与的显著性。这些 P 值以前被认为是独立的。在这里,我们表明情况并非如此,许多途径可以通过“串扰”现象极大地影响彼此的 P 值。尽管各种途径相互影响是直观的,但该现象的存在和程度尚未得到严格研究,最重要的是,目前没有可用的技术能够量化这种串扰的程度。在这里,我们表明,三种主要的途径分析方法类别(富集分析、功能分类评分和基于拓扑的方法)都受到串扰现象的严重影响。使用真实的途径和数据,我们表明在某些情况下,具有显著 P 值的途径在生物学上没有意义,而当去除其他途径的串扰影响时,一些具有非显著 P 值的生物学上有意义的途径在统计学上变得显著。我们描述了一种能够检测、量化和纠正串扰效应以及识别独立功能模块的技术。我们在涉及三种表型和两种物种的四个实验的数据上评估了这种新方法。该方法有望更好地理解个体实验结果,并对特定表型的现有信号通路进行更精细的定义。