Albrecht-Kossel-Institute for Neuroregeneration, Centre for Mental Health, University of Rostock, Rostock, Germany.
PLoS Genet. 2013;9(8):e1003632. doi: 10.1371/journal.pgen.1003632. Epub 2013 Aug 1.
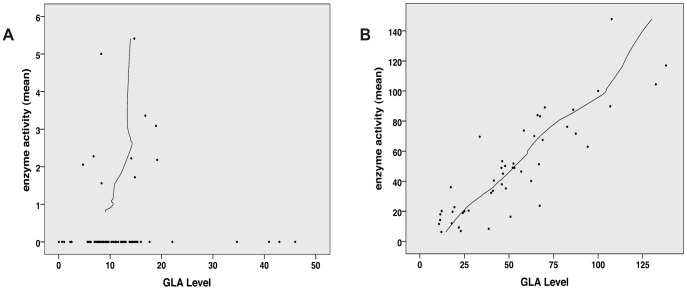
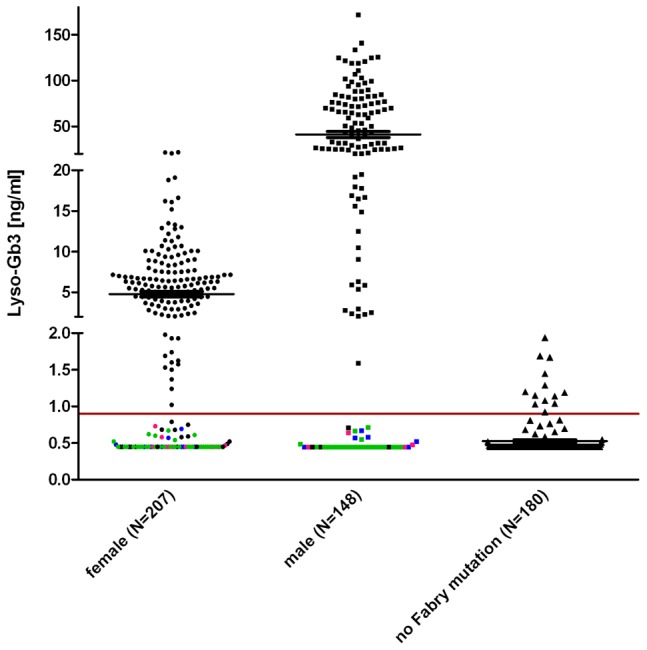
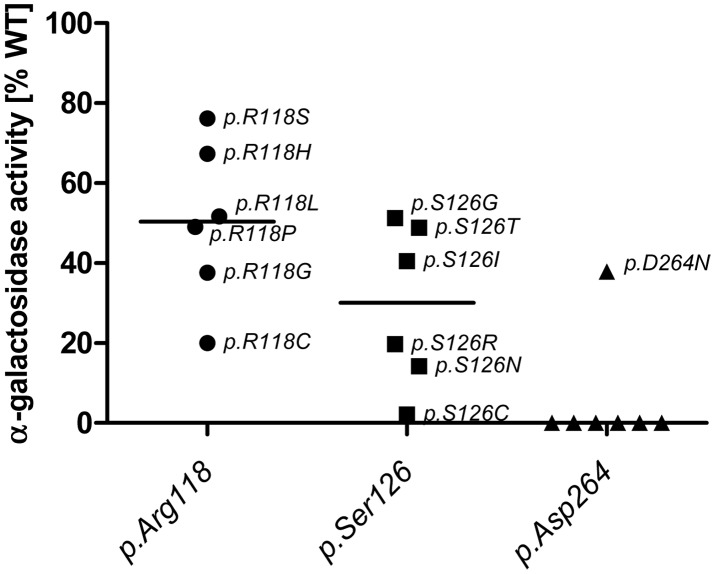
Fabry disease (FD) is an X-linked hereditary defect of glycosphingolipid storage caused by mutations in the gene encoding the lysosomal hydrolase α-galactosidase A (GLA, α-gal A). To date, over 400 mutations causing amino acid substitutions have been described. Most of these mutations are related to the classical Fabry phenotype. Generally in lysosomal storage disorders a reliable genotype/phenotype correlation is difficult to achieve, especially in FD with its X-linked mode of inheritance. In order to predict the metabolic consequence of a given mutation, we combined in vitro enzyme activity with in vivo biomarker data. Furthermore, we used the pharmacological chaperone (PC) 1-deoxygalactonojirimycin (DGJ) as a tool to analyse the influence of individual mutations on subcellular organelle-trafficking and stability. We analysed a significant number of mutations and correlated the obtained properties to the clinical manifestation related to the mutation in order to improve our knowledge of the identity of functional relevant amino acids. Additionally, we illustrate the consequences of different mutations on plasma lyso-globotriaosylsphingosine (lyso-Gb3) accumulation in the patients' plasma, a biomarker proven to reflect the impaired substrate clearance caused by specific mutations. The established system enables us to provide information for the clinical relevance of PC therapy for a given mutant. Finally, in order to generate reliable predictions of mutant GLA defects we compared the different data sets to reveal the most coherent system to reflect the clinical situation.
法布里病(FD)是一种 X 连锁遗传性糖脂贮积病,由编码溶酶体水解酶α-半乳糖苷酶 A(GLA,α-gal A)的基因突变引起。迄今为止,已经描述了超过 400 种导致氨基酸取代的突变。这些突变大多数与经典的法布里表型有关。一般来说,在溶酶体贮积病中,很难实现可靠的基因型/表型相关性,尤其是在 X 连锁遗传方式的 FD 中。为了预测给定突变的代谢后果,我们将体外酶活性与体内生物标志物数据相结合。此外,我们使用药理学伴侣(PC)1-脱氧半乳糖基氮杂环庚烷(DGJ)作为工具来分析个体突变对亚细胞器运输和稳定性的影响。我们分析了大量的突变,并将获得的特性与与突变相关的临床表现相关联,以提高我们对功能相关氨基酸的认识。此外,我们还说明了不同突变对患者血浆中溶神经酰基鞘氨醇(lyso-Gb3)积累的影响,该生物标志物被证明反映了特定突变导致的底物清除受损。所建立的系统使我们能够为特定突变体的 PC 治疗的临床相关性提供信息。最后,为了对突变 GLA 缺陷产生可靠的预测,我们比较了不同数据集,以揭示最能反映临床情况的系统。