Roslyn and Leslie Goldstein Laboratory for Stem Cell Biology and Regenerative Medicine, Albert Einstein College of Medicine, Bronx, New York, USA.
PLoS One. 2013 Aug 13;8(8):e72698. doi: 10.1371/journal.pone.0072698. eCollection 2013.
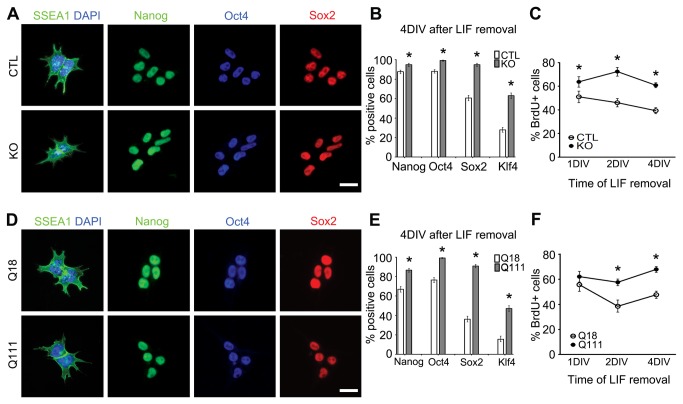
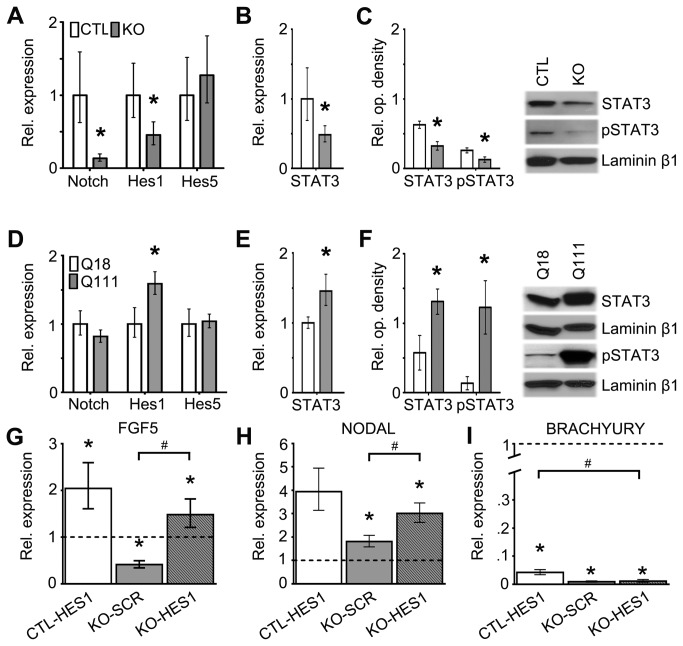
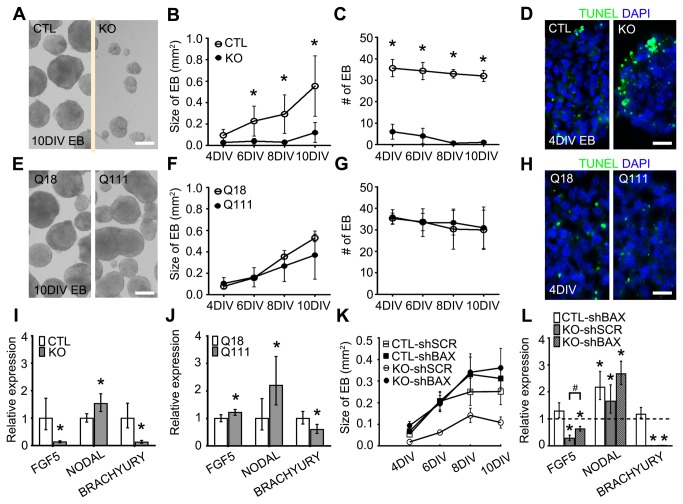
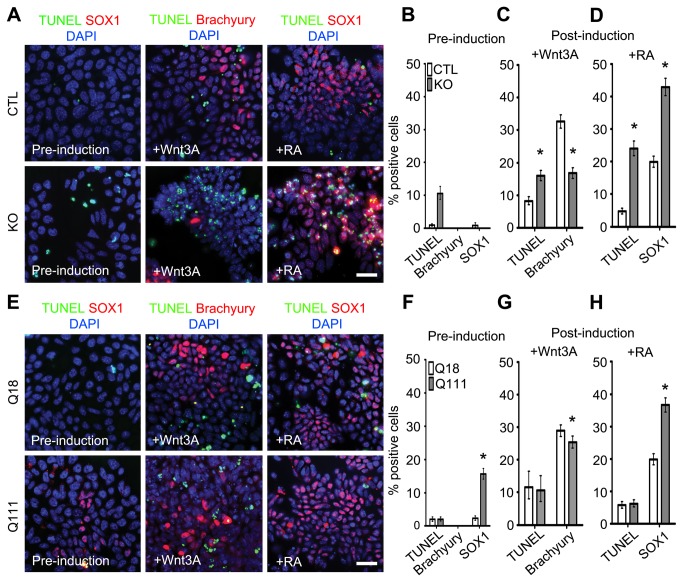
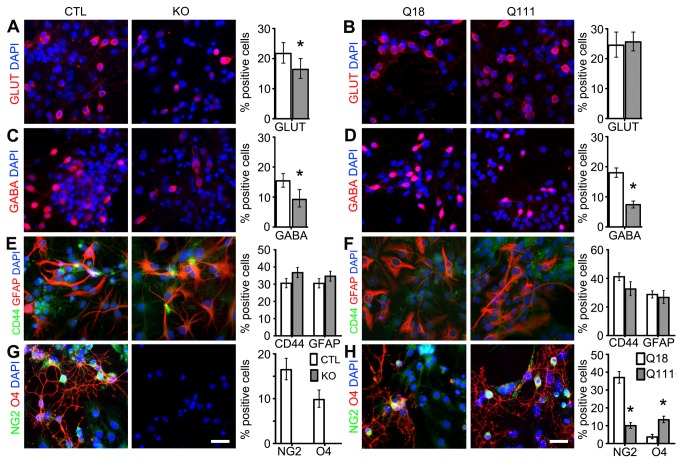
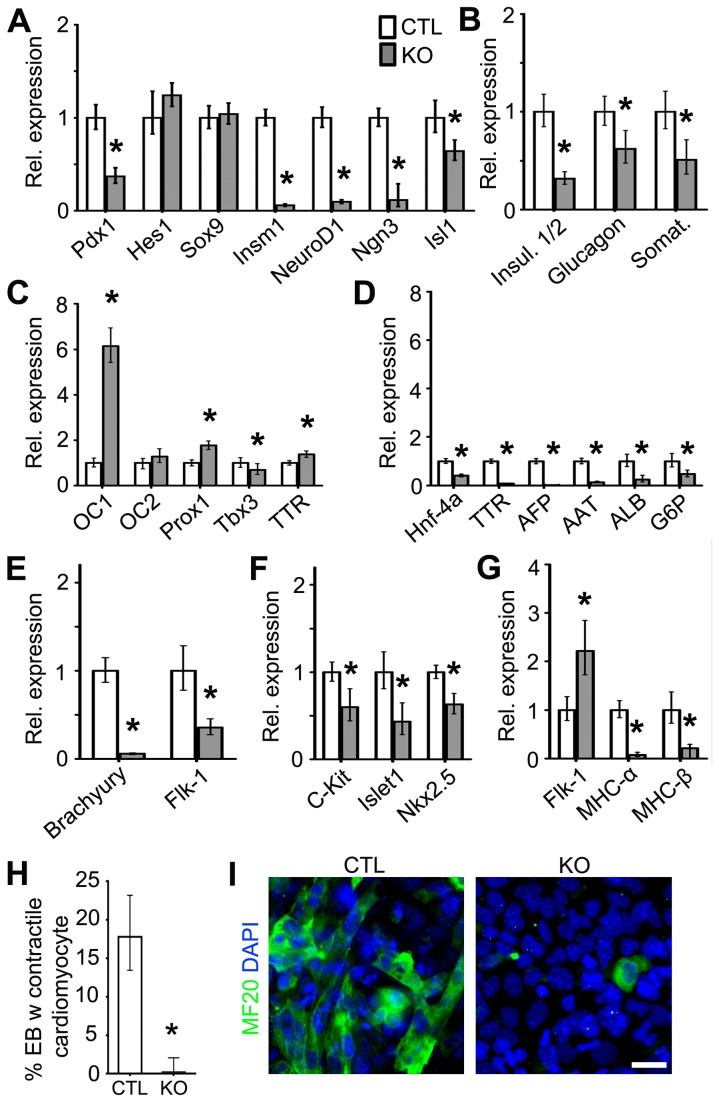
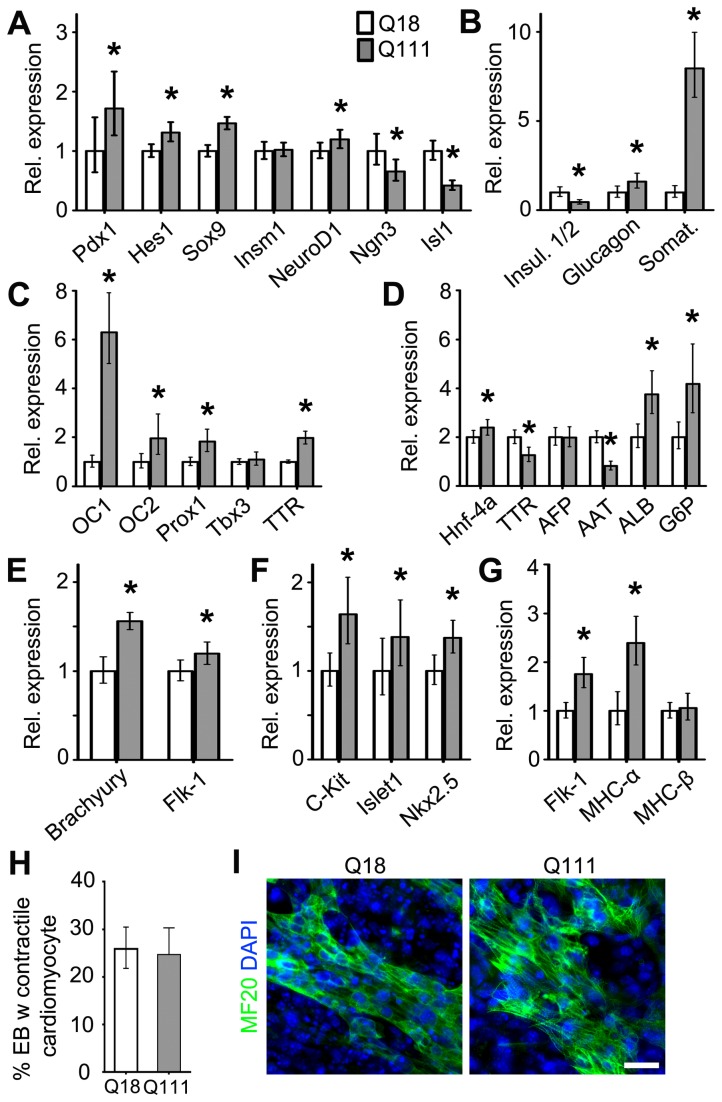
Huntington's disease (HD) is a neurodegenerative disease caused by abnormal polyglutamine expansion in the huntingtin protein (Htt). Although both Htt and the HD pathogenic mutation (mHtt) are implicated in early developmental events, their individual involvement has not been adequately explored. In order to better define the developmental functions and pathological consequences of the normal and mutant proteins, respectively, we employed embryonic stem cell (ESC) expansion, differentiation and induction experiments using huntingtin knock-out (KO) and mutant huntingtin knock-in (Q111) mouse ESC lines. In KO ESCs, we observed impairments in the spontaneous specification and survival of ectodermal and mesodermal lineages during embryoid body formation and under inductive conditions using retinoic acid and Wnt3A, respectively. Ablation of BAX improves cell survival, but failed to correct defects in germ layer specification. In addition, we observed ensuing impairments in the specification and maturation of neural, hepatic, pancreatic and cardiomyocyte lineages. These developmental deficits occurred in concert with alterations in Notch, Hes1 and STAT3 signaling pathways. Moreover, in Q111 ESCs, we observed differential developmental stage-specific alterations in lineage specification and maturation. We also observed changes in Notch/STAT3 expression and activation. Our observations underscore essential roles of Htt in the specification of ectoderm, endoderm and mesoderm, in the specification of neural and non-neural organ-specific lineages, as well as cell survival during early embryogenesis. Remarkably, these developmental events are differentially deregulated by mHtt, raising the possibility that HD-associated early developmental impairments may contribute not only to region-specific neurodegeneration, but also to non-neural co-morbidities.
亨廷顿病(HD)是一种由亨廷顿蛋白(Htt)中异常的多聚谷氨酰胺扩展引起的神经退行性疾病。尽管 Htt 和 HD 致病突变(mHtt)都与早期发育事件有关,但它们各自的参与尚未得到充分探索。为了更好地定义正常和突变蛋白的发育功能和病理后果,我们分别使用亨廷顿敲除(KO)和突变亨廷顿敲入(Q111)小鼠胚胎干细胞(ESC)系进行 ESC 扩增、分化和诱导实验。在 KO ESC 中,我们观察到在胚状体形成过程中和在使用视黄酸和 Wnt3A 的诱导条件下,外胚层和中胚层谱系的自发特化和存活受损。BAX 的消融改善了细胞存活,但未能纠正胚层特化缺陷。此外,我们观察到神经、肝、胰腺和心肌细胞谱系的特化和成熟缺陷。这些发育缺陷与 Notch、Hes1 和 STAT3 信号通路的改变同时发生。此外,在 Q111 ESC 中,我们观察到谱系特化和成熟的不同发育阶段特异性改变。我们还观察到 Notch/STAT3 表达和激活的变化。我们的观察结果强调了 Htt 在外胚层、内胚层和中胚层的特化、神经和非神经器官特异性谱系的特化以及早期胚胎发生期间细胞存活中的重要作用。值得注意的是,mHtt 导致这些发育事件的差异调节,这增加了 HD 相关早期发育损伤不仅可能导致区域特异性神经退行性变,而且可能导致非神经共病的可能性。