Laboratoire de Physique Statistique de l'Ecole Normale Supérieure - UMR 8550, associé au CNRS et à l'Université Pierre et Marie Curie, Paris, France.
PLoS Comput Biol. 2013;9(8):e1003176. doi: 10.1371/journal.pcbi.1003176. Epub 2013 Aug 22.
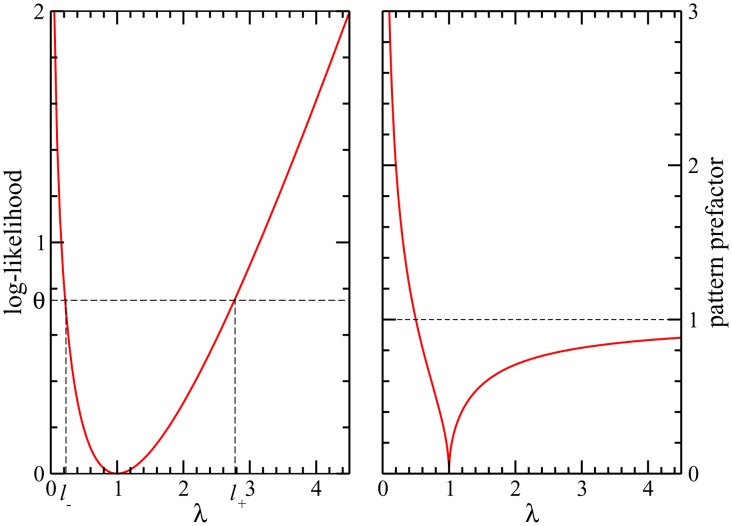
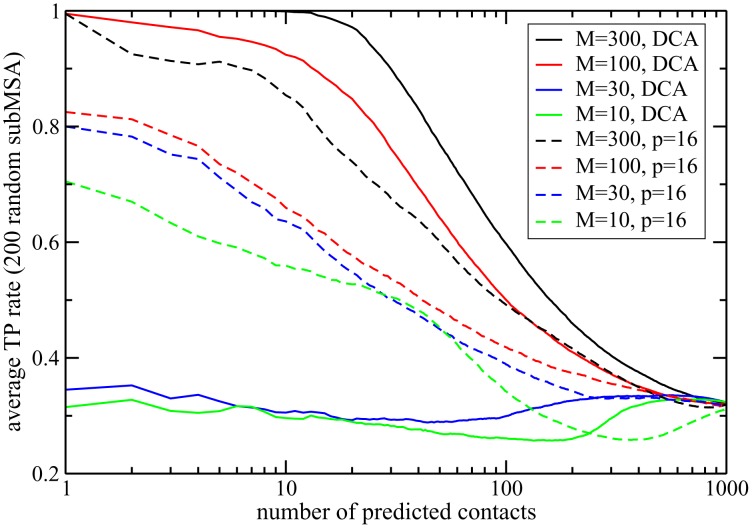
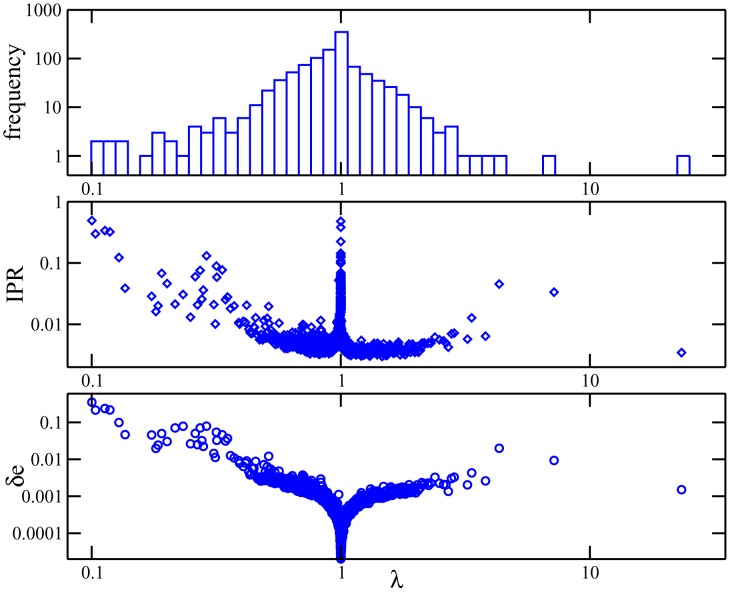
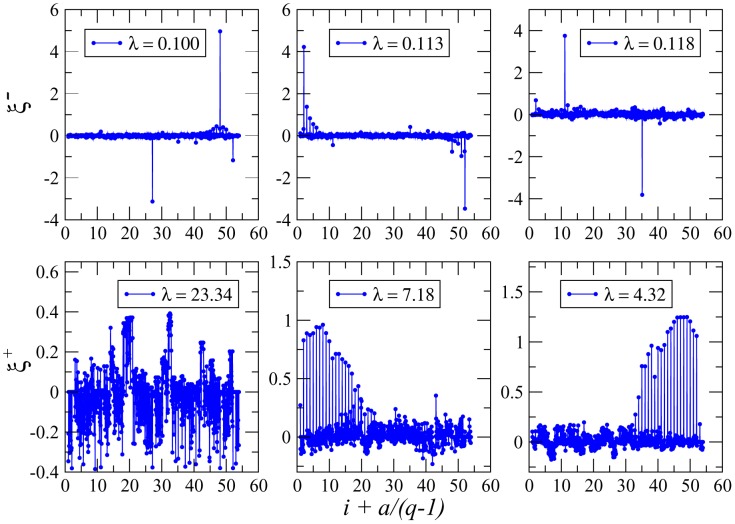
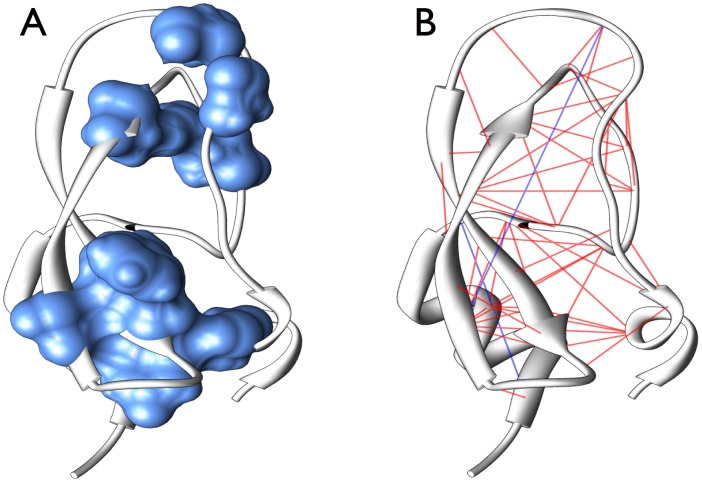
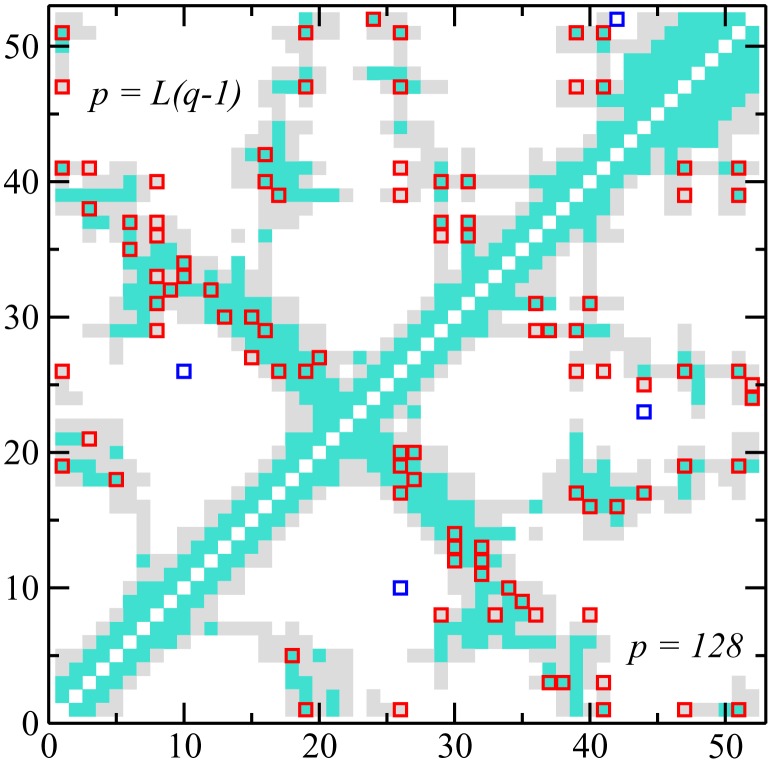
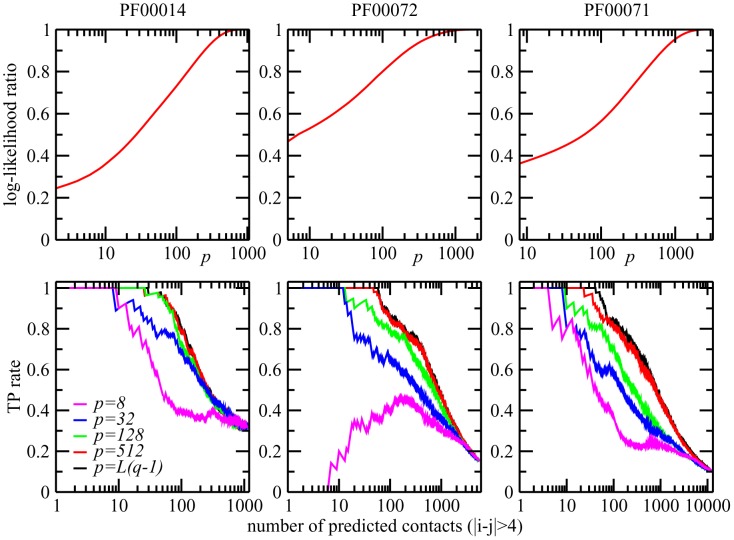
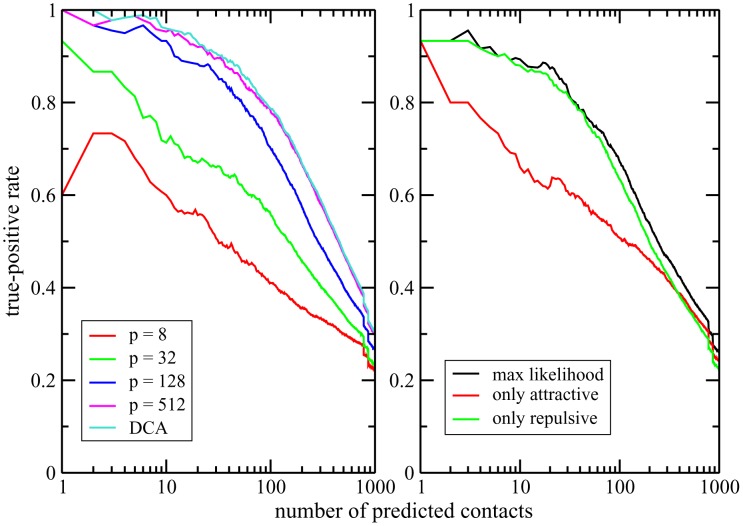
Various approaches have explored the covariation of residues in multiple-sequence alignments of homologous proteins to extract functional and structural information. Among those are principal component analysis (PCA), which identifies the most correlated groups of residues, and direct coupling analysis (DCA), a global inference method based on the maximum entropy principle, which aims at predicting residue-residue contacts. In this paper, inspired by the statistical physics of disordered systems, we introduce the Hopfield-Potts model to naturally interpolate between these two approaches. The Hopfield-Potts model allows us to identify relevant 'patterns' of residues from the knowledge of the eigenmodes and eigenvalues of the residue-residue correlation matrix. We show how the computation of such statistical patterns makes it possible to accurately predict residue-residue contacts with a much smaller number of parameters than DCA. This dimensional reduction allows us to avoid overfitting and to extract contact information from multiple-sequence alignments of reduced size. In addition, we show that low-eigenvalue correlation modes, discarded by PCA, are important to recover structural information: the corresponding patterns are highly localized, that is, they are concentrated in few sites, which we find to be in close contact in the three-dimensional protein fold.
多种方法已经探索了同源蛋白多重序列比对中残基的共变,以提取功能和结构信息。其中包括主成分分析(PCA),它可以识别相关性最强的残基组,以及直接耦合分析(DCA),这是一种基于最大熵原理的全局推断方法,旨在预测残基-残基接触。在本文中,受无序系统统计物理的启发,我们引入了 Hopfield-Potts 模型,将这两种方法自然地结合起来。Hopfield-Potts 模型使我们能够从残基-残基相关矩阵的本征模和本征值的知识中识别相关的“模式”。我们展示了如何计算这些统计模式,以便用比 DCA 少得多的参数准确地预测残基-残基接触。这种降维使得我们可以避免过度拟合,并从较小尺寸的多重序列比对中提取接触信息。此外,我们还表明,PCA 丢弃的低本征值相关模式对于恢复结构信息非常重要:对应的模式高度局域化,即它们集中在少数几个位点,我们发现这些位点在三维蛋白质折叠中彼此非常接近。