Center for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China.
BNLMS, Peking-Tsinghua Center for Life Sciences at the College of Chemistry and Molecular Engineering, Peking University, Beijing, China.
Elife. 2023 Feb 17;12:e81850. doi: 10.7554/eLife.81850.
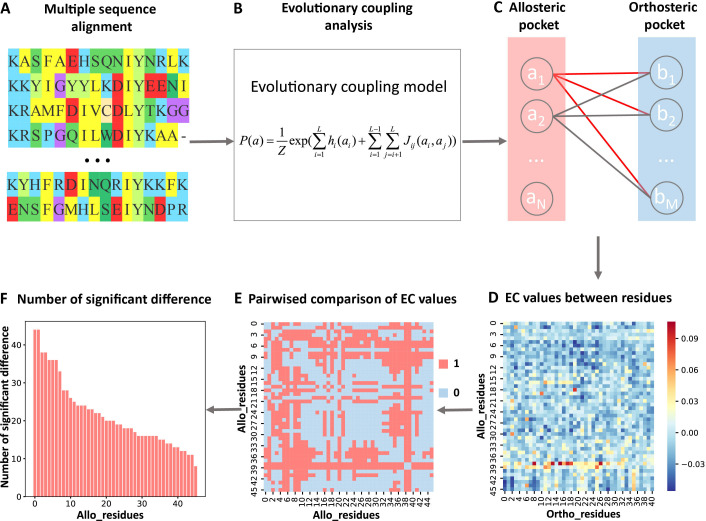
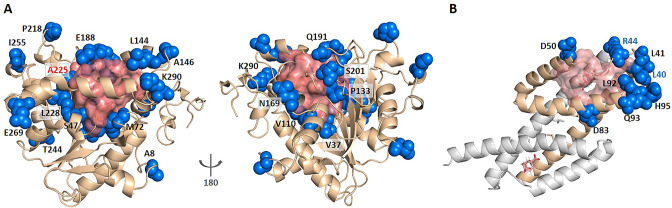
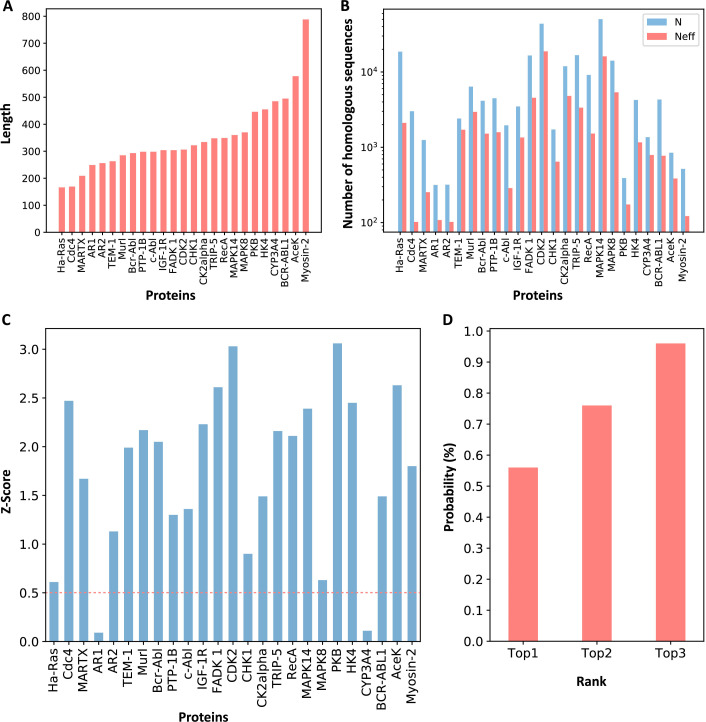
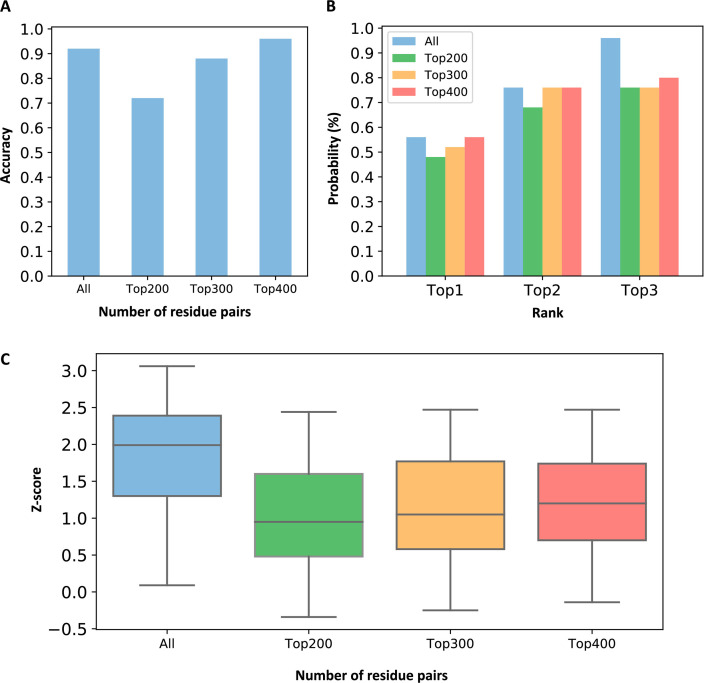
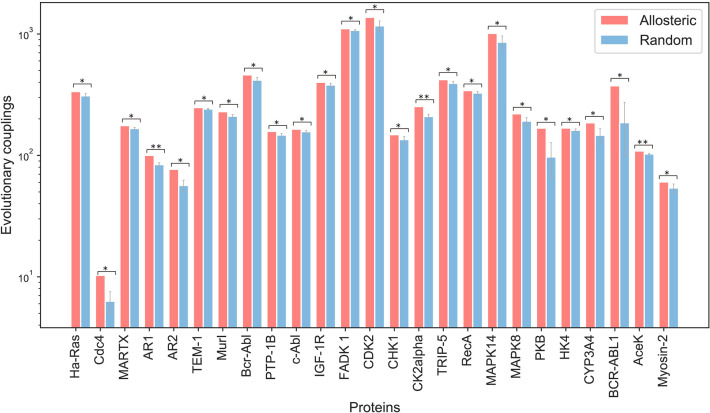
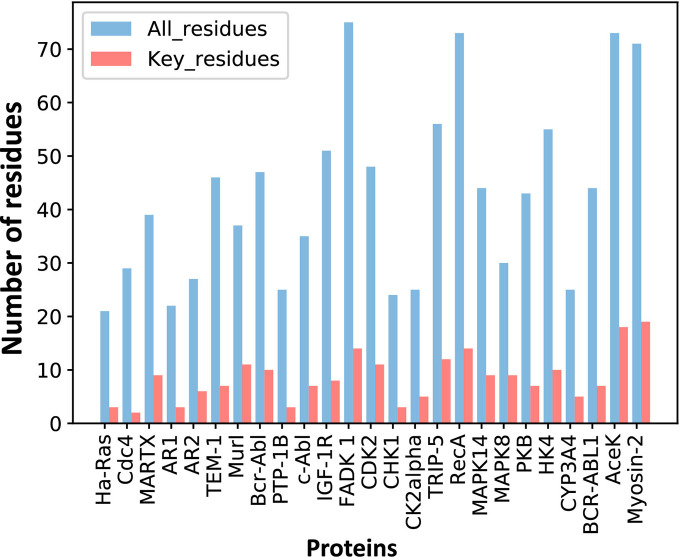
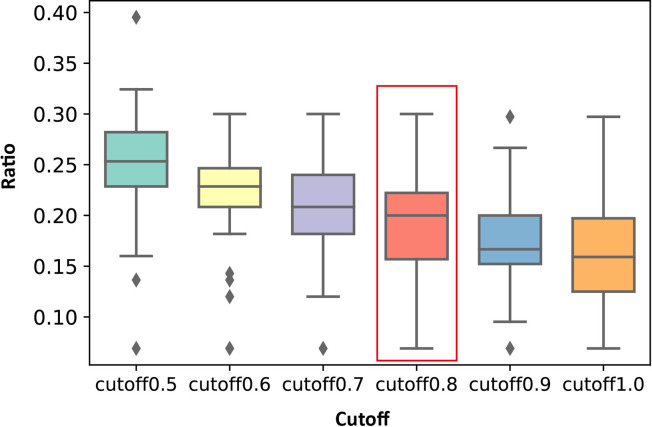
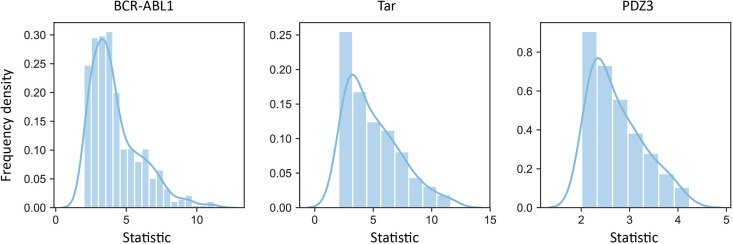
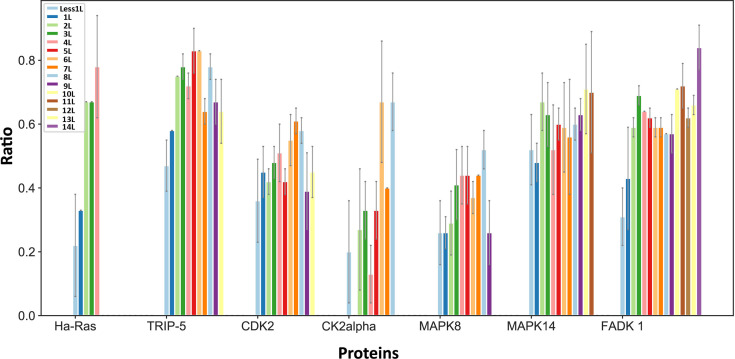
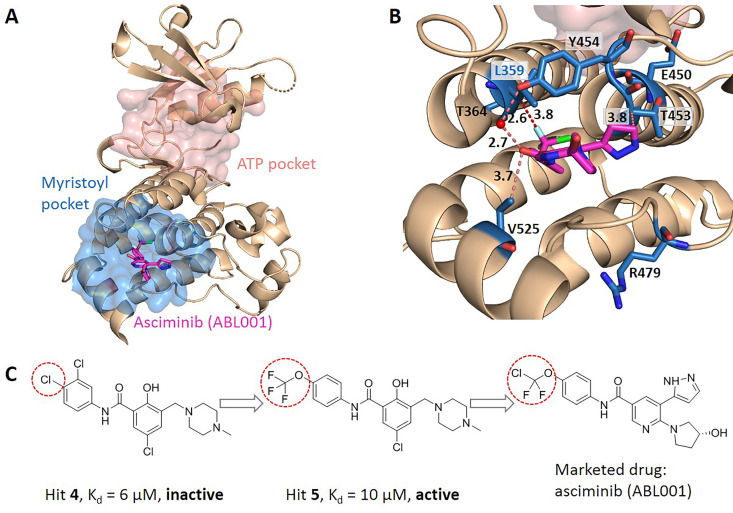
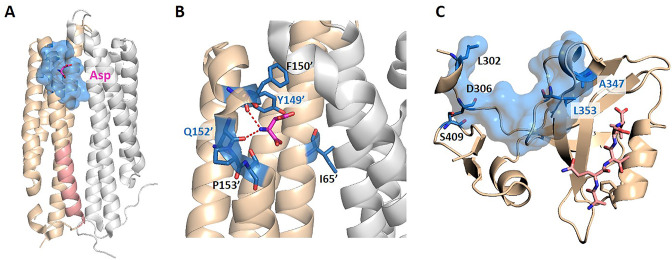
Allostery is fundamental to many biological processes. Due to the distant regulation nature, how allosteric mutations, modifications, and effector binding impact protein function is difficult to forecast. In protein engineering, remote mutations cannot be rationally designed without large-scale experimental screening. Allosteric drugs have raised much attention due to their high specificity and possibility of overcoming existing drug-resistant mutations. However, optimization of allosteric compounds remains challenging. Here, we developed a novel computational method KeyAlloSite to predict allosteric site and to identify key allosteric residues (allo-residues) based on the evolutionary coupling model. We found that protein allosteric sites are strongly coupled to orthosteric site compared to non-functional sites. We further inferred key allo-residues by pairwise comparing the difference of evolutionary coupling scores of each residue in the allosteric pocket with the functional site. Our predicted key allo-residues are in accordance with previous experimental studies for typical allosteric proteins like BCR-ABL1, Tar, and PDZ3, as well as key cancer mutations. We also showed that KeyAlloSite can be used to predict key allosteric residues distant from the catalytic site that are important for enzyme catalysis. Our study demonstrates that weak coevolutionary couplings contain important information of protein allosteric regulation function. KeyAlloSite can be applied in studying the evolution of protein allosteric regulation, designing and optimizing allosteric drugs, and performing functional protein design and enzyme engineering.
变构作用是许多生物过程的基础。由于变构调控的本质,变构突变、修饰和效应物结合如何影响蛋白质功能是难以预测的。在蛋白质工程中,如果没有大规模的实验筛选,远程突变就无法进行合理设计。变构药物由于其高特异性和克服现有耐药突变的可能性而引起了广泛关注。然而,变构化合物的优化仍然具有挑战性。在这里,我们开发了一种新的计算方法 KeyAlloSite,该方法基于进化耦联模型来预测变构位点和识别关键变构残基(allo-residues)。我们发现与非功能位点相比,蛋白质变构位点与正构位点的耦联要强得多。我们通过比较变构口袋中每个残基的进化耦联分数与功能位点之间的差异,进一步推断关键 allo-residues。我们预测的关键 allo-residues与典型变构蛋白(如 BCR-ABL1、Tar 和 PDZ3)以及关键癌症突变的先前实验研究相符。我们还表明,KeyAlloSite 可用于预测远离催化位点的关键变构残基,这些残基对于酶催化非常重要。我们的研究表明,弱共进化耦联包含蛋白质变构调控功能的重要信息。KeyAlloSite 可应用于研究蛋白质变构调控的进化、设计和优化变构药物以及进行功能蛋白设计和酶工程。