Adhikari Badri, Hou Jie, Cheng Jianlin
Department of Mathematics and Computer Science, University of Missouri-St. Louis, St. Louis, Missouri.
Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri.
Proteins. 2018 Mar;86 Suppl 1(Suppl 1):84-96. doi: 10.1002/prot.25405. Epub 2017 Oct 31.
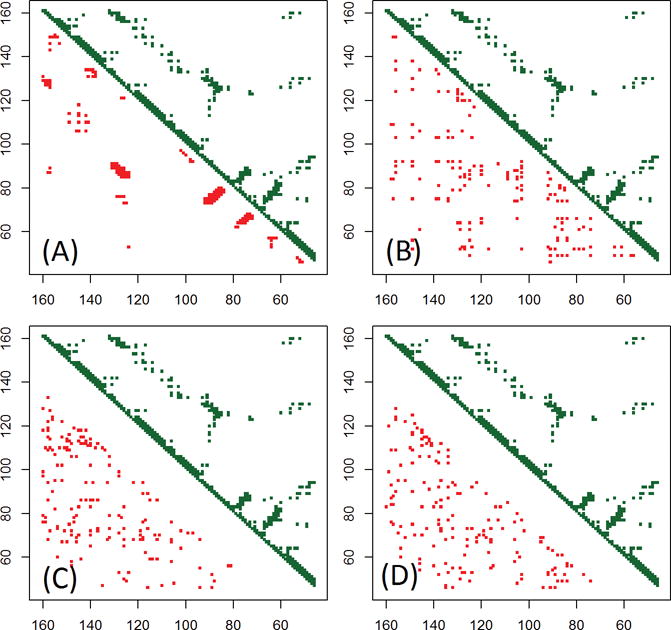
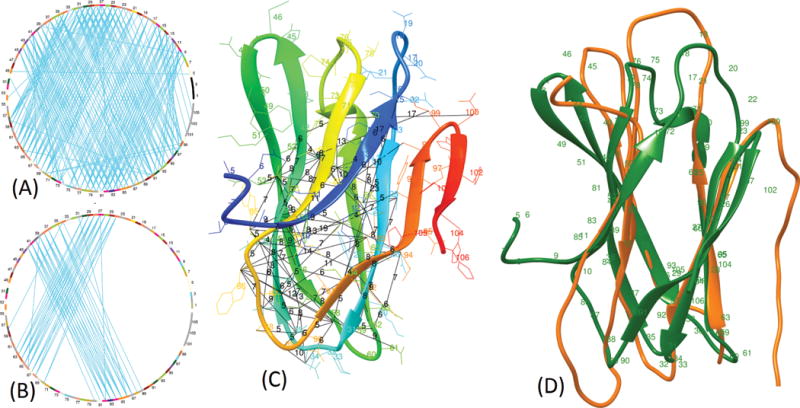
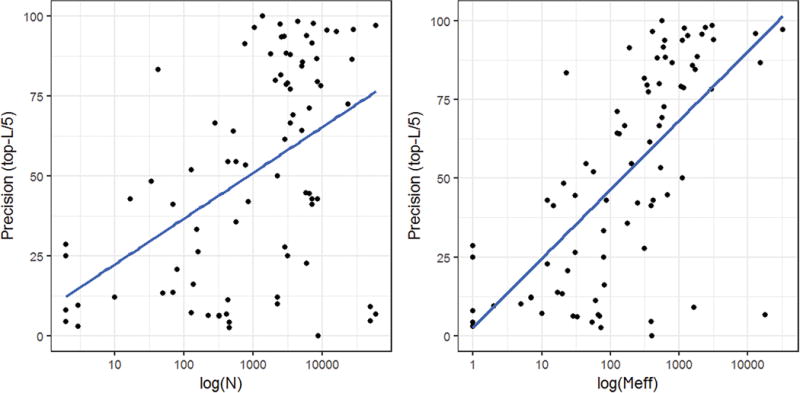
In this study, we report the evaluation of the residue-residue contacts predicted by our three different methods in the CASP12 experiment, focusing on studying the impact of multiple sequence alignment, residue coevolution, and machine learning on contact prediction. The first method (MULTICOM-NOVEL) uses only traditional features (sequence profile, secondary structure, and solvent accessibility) with deep learning to predict contacts and serves as a baseline. The second method (MULTICOM-CONSTRUCT) uses our new alignment algorithm to generate deep multiple sequence alignment to derive coevolution-based features, which are integrated by a neural network method to predict contacts. The third method (MULTICOM-CLUSTER) is a consensus combination of the predictions of the first two methods. We evaluated our methods on 94 CASP12 domains. On a subset of 38 free-modeling domains, our methods achieved an average precision of up to 41.7% for top L/5 long-range contact predictions. The comparison of the three methods shows that the quality and effective depth of multiple sequence alignments, coevolution-based features, and machine learning integration of coevolution-based features and traditional features drive the quality of predicted protein contacts. On the full CASP12 dataset, the coevolution-based features alone can improve the average precision from 28.4% to 41.6%, and the machine learning integration of all the features further raises the precision to 56.3%, when top L/5 predicted long-range contacts are evaluated. And the correlation between the precision of contact prediction and the logarithm of the number of effective sequences in alignments is 0.66.
在本研究中,我们报告了在蛋白质结构预测技术关键评估第12轮(CASP12)实验中,对我们三种不同方法预测的残基-残基接触的评估,重点是研究多序列比对、残基协同进化和机器学习对接触预测的影响。第一种方法(MULTICOM-NOVEL)仅使用传统特征(序列谱、二级结构和溶剂可及性)结合深度学习来预测接触,并作为基线。第二种方法(MULTICOM-CONSTRUCT)使用我们新的比对算法生成深度多序列比对,以得出基于协同进化的特征,这些特征通过神经网络方法进行整合以预测接触。第三种方法(MULTICOM-CLUSTER)是前两种方法预测结果的一致性组合。我们在94个CASP12结构域上评估了我们的方法。在38个自由建模结构域的子集上,对于前L/5个长程接触预测,我们的方法平均精度高达41.7%。三种方法的比较表明,多序列比对的质量和有效深度、基于协同进化的特征以及基于协同进化特征与传统特征的机器学习整合,推动了预测蛋白质接触的质量。在完整的CASP12数据集上,当评估前L/5个预测的长程接触时,仅基于协同进化的特征就能将平均精度从28.4%提高到41.6%,所有特征的机器学习整合进一步将精度提高到56.3%。并且接触预测精度与比对中有效序列数量的对数之间的相关性为0.66。