Department of Biochemistry, Carver College of Medicine, University of Iowa , Iowa City, IA , USA.
Crit Rev Biochem Mol Biol. 2013 Nov-Dec;48(6):561-74. doi: 10.3109/10409238.2013.838204. Epub 2013 Sep 19.
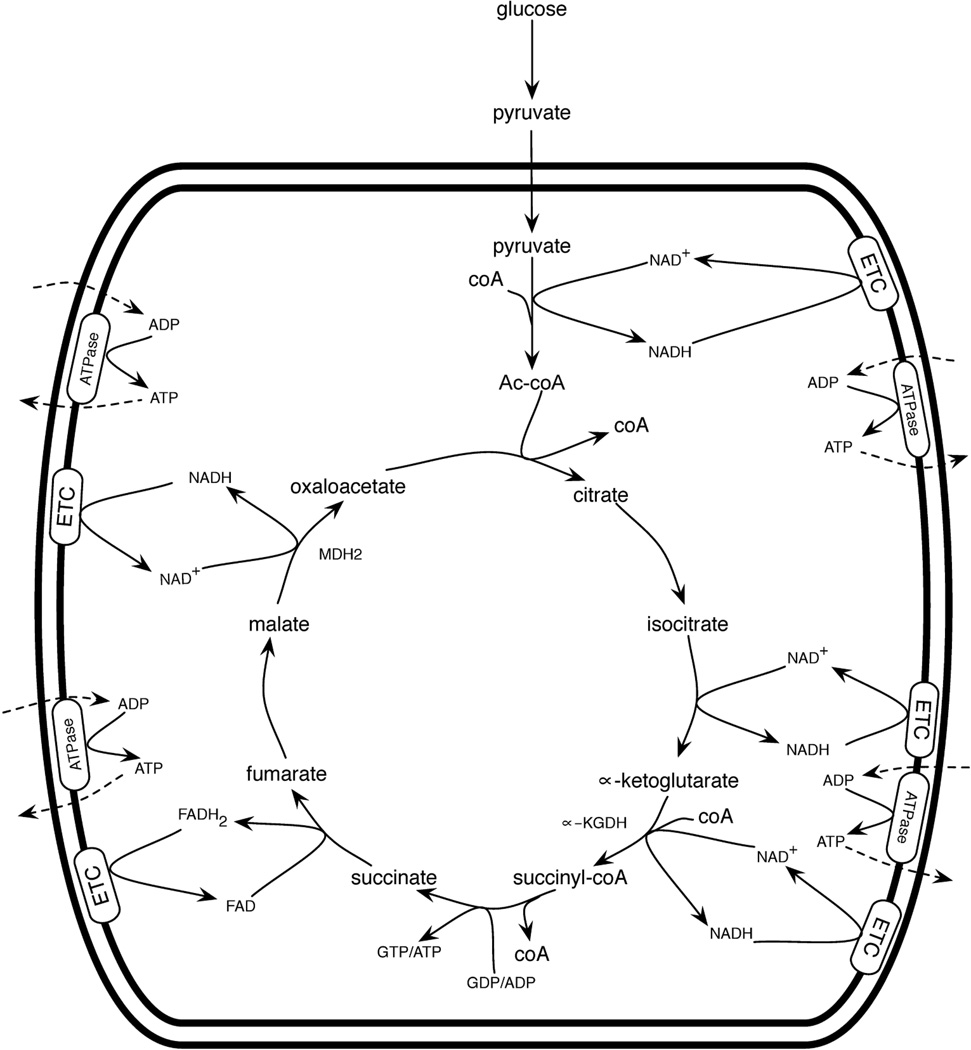
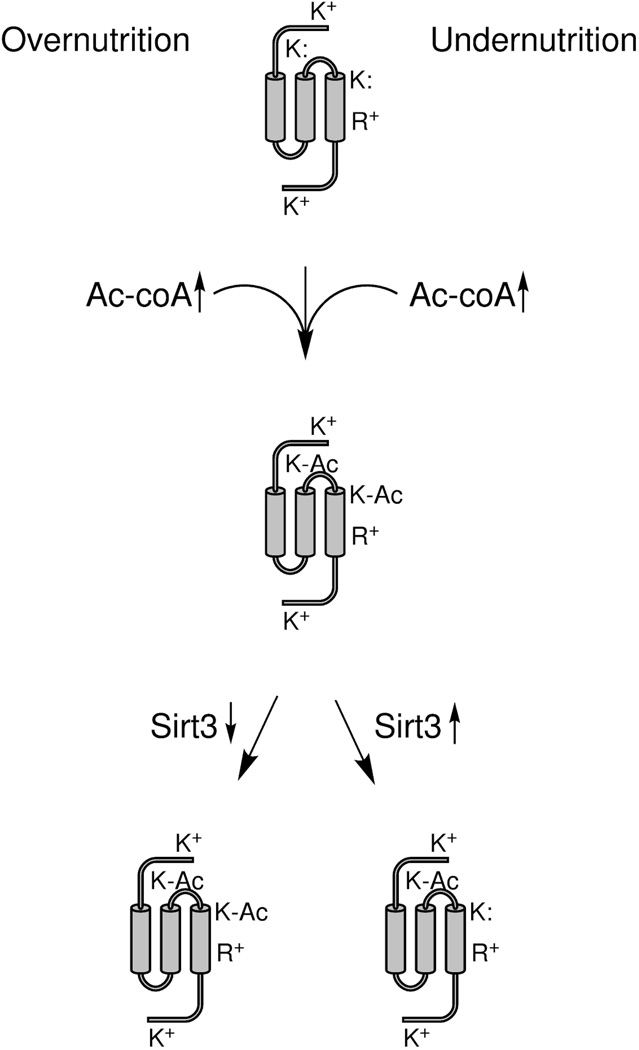
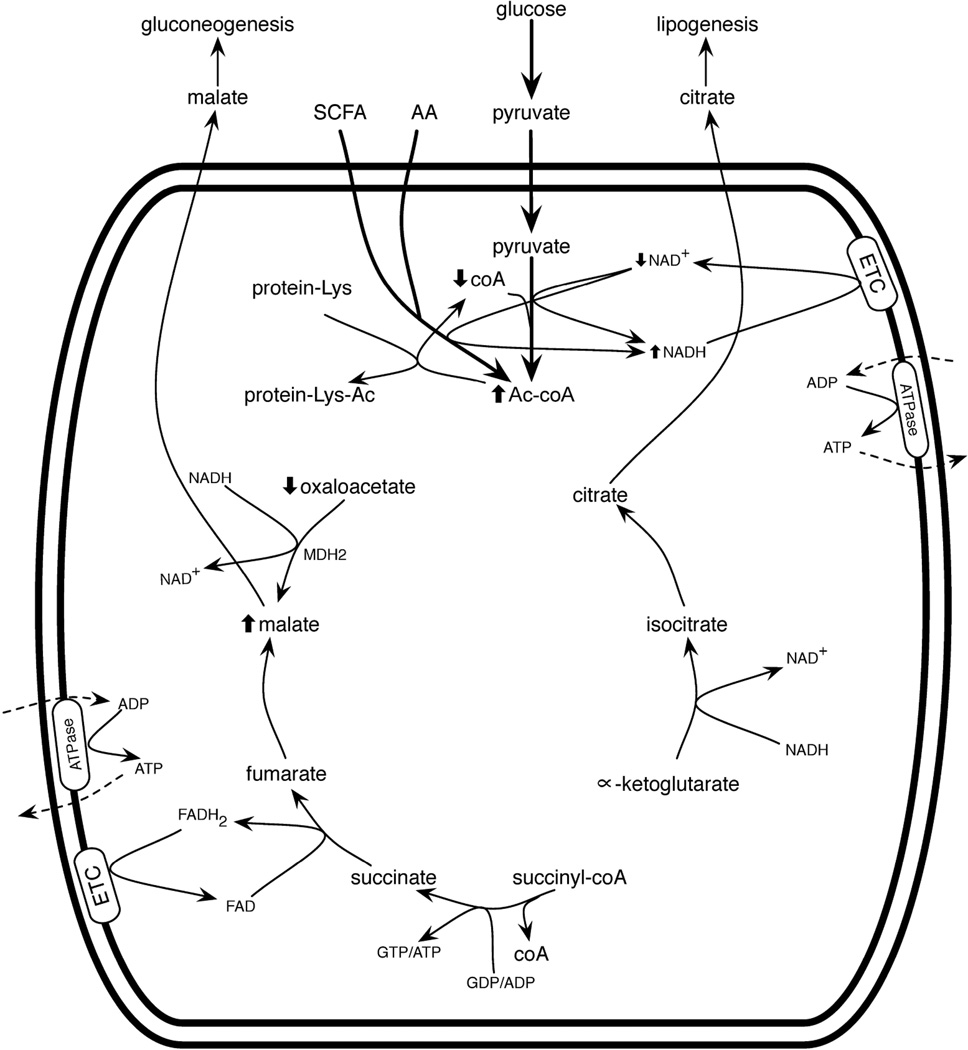
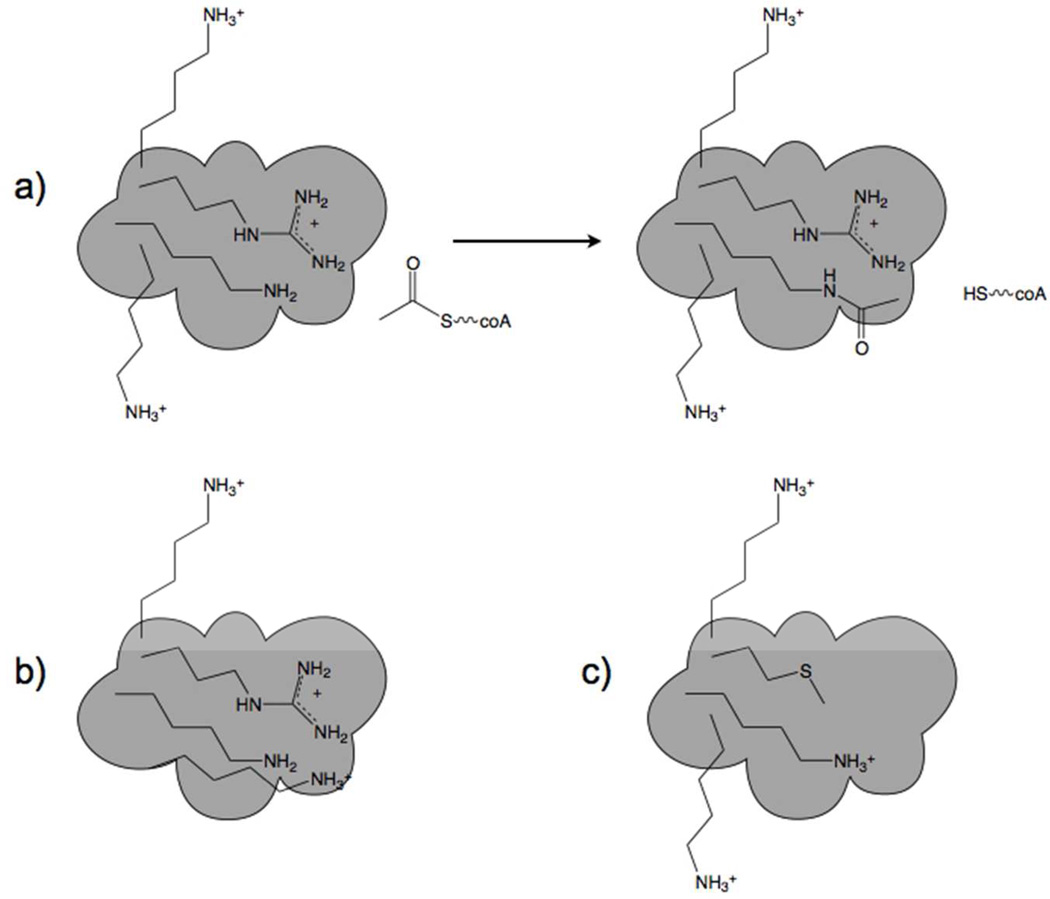
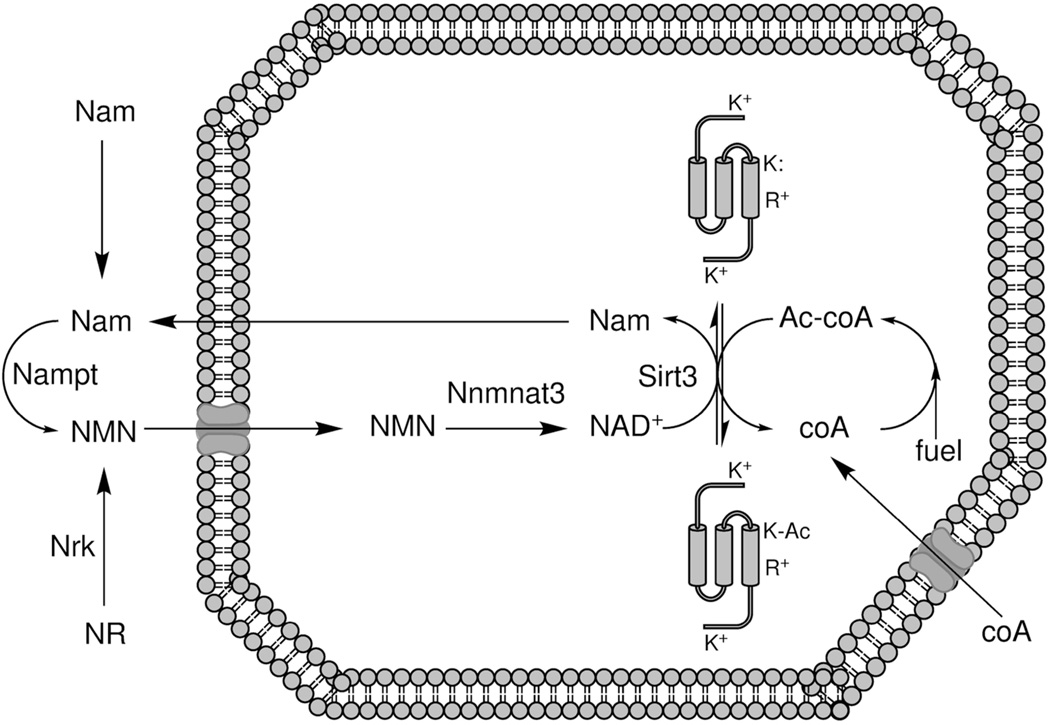
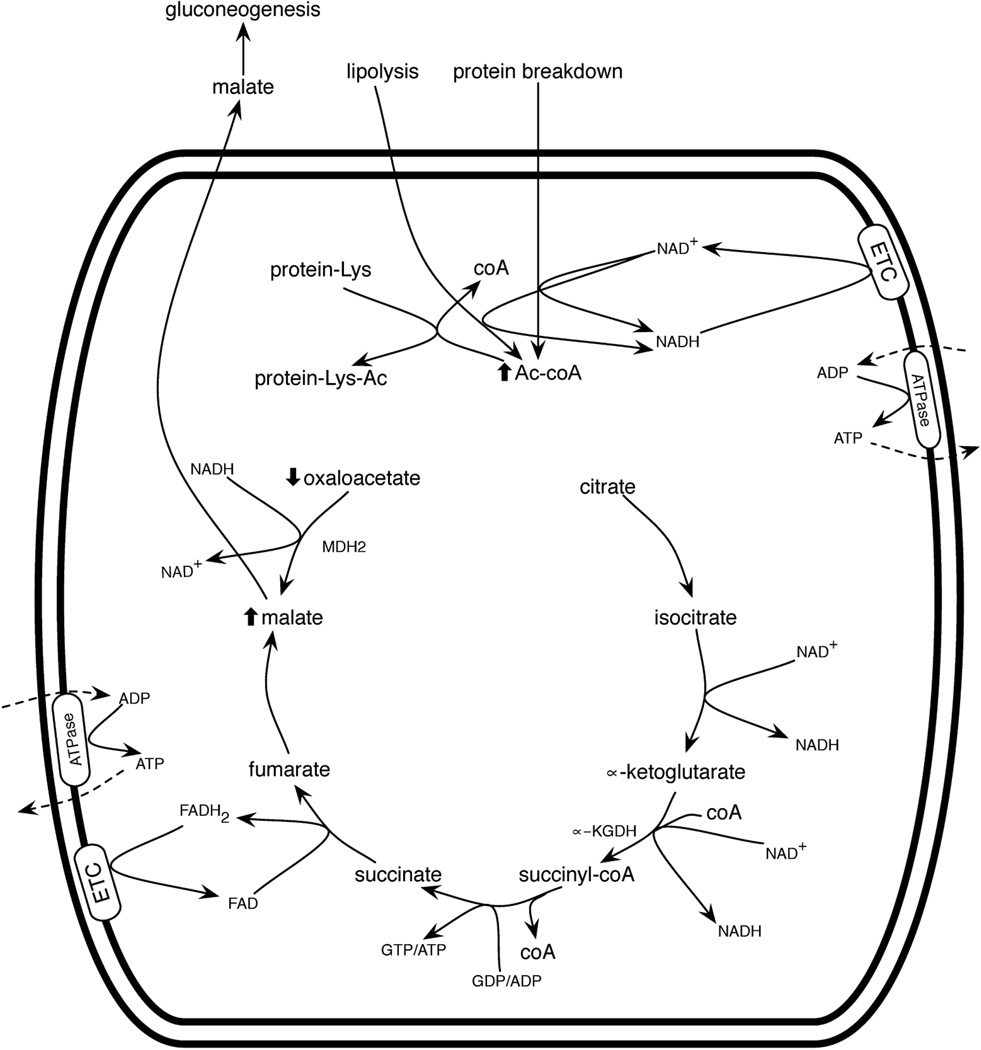
Hormone systems evolved over 500 million years of animal natural history to motivate feeding behavior and convert excess calories to fat. These systems produced vertebrates, including humans, who are famine-resistant but sensitive to obesity in environments of persistent overnutrition. We looked for cell-intrinsic metabolic features, which might have been subject to an evolutionary drive favoring lipogenesis. Mitochondrial protein acetylation appears to be such a system. Because mitochondrial acetyl-coA is the central mediator of fuel oxidation and is saturable, this metabolite is postulated to be the fundamental indicator of energy excess, which imprints a memory of nutritional imbalances by covalent modification. Fungal and invertebrate mitochondria have highly acetylated mitochondrial proteomes without an apparent mitochondrially targeted protein lysine acetyltransferase. Thus, mitochondrial acetylation is hypothesized to have evolved as a nonenzymatic phenomenon. Because the pKa of a nonperturbed Lys is 10.4 and linkage of a carbonyl carbon to an ε amino group cannot be formed with a protonated Lys, we hypothesize that acetylation occurs on residues with depressed pKa values, accounting for the propensity of acetylation to hit active sites and suggesting that regulatory Lys residues may have been under selective pressure to avoid or attract acetylation throughout animal evolution. In addition, a shortage of mitochondrial oxaloacetate under ketotic conditions can explain why macronutrient insufficiency also produces mitochondrial hyperacetylation. Reduced mitochondrial activity during times of overnutrition and undernutrition would improve fitness by virtue of resource conservation. Micronutrient insufficiency is predicted to exacerbate mitochondrial hyperacetylation. Nicotinamide riboside and Sirt3 activity are predicted to relieve mitochondrial inhibition.
激素系统在 5 亿多年的动物自然史中进化,以激发进食行为并将多余的卡路里转化为脂肪。这些系统产生了脊椎动物,包括人类,人类具有抗饥饿能力,但在持续营养过剩的环境中容易肥胖。我们寻找细胞内在的代谢特征,这些特征可能受到促进脂肪生成的进化驱动力的影响。线粒体蛋白乙酰化似乎就是这样一个系统。由于线粒体乙酰辅酶 A 是燃料氧化的核心调节剂,并且是饱和的,因此该代谢物被假设为能量过剩的基本指标,通过共价修饰留下营养失衡的记忆。真菌和无脊椎动物的线粒体具有高度乙酰化的线粒体蛋白质组,而没有明显的靶向线粒体的蛋白赖氨酸乙酰转移酶。因此,线粒体乙酰化被假设为一种非酶促现象而进化。由于未受干扰的赖氨酸的 pKa 值为 10.4,并且羰基碳与 ε 氨基基团的连接不能与质子化的赖氨酸形成,因此我们假设乙酰化发生在 pKa 值降低的残基上,这解释了乙酰化倾向于击中活性位点的原因,并表明调节赖氨酸残基可能在整个动物进化过程中一直受到避免或吸引乙酰化的选择性压力。此外,酮症条件下线粒体草酰乙酸的短缺可以解释为什么宏量营养素不足也会导致线粒体过度乙酰化。在营养过剩和营养不足时减少线粒体活性会通过资源节约来提高适应性。预测微量营养素不足会加剧线粒体过度乙酰化。烟酰胺核苷和 Sirt3 活性被预测可以缓解线粒体抑制。