Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, Washington, United States of America.
PLoS Biol. 2013 Sep;11(9):e1001661. doi: 10.1371/journal.pbio.1001661. Epub 2013 Sep 17.
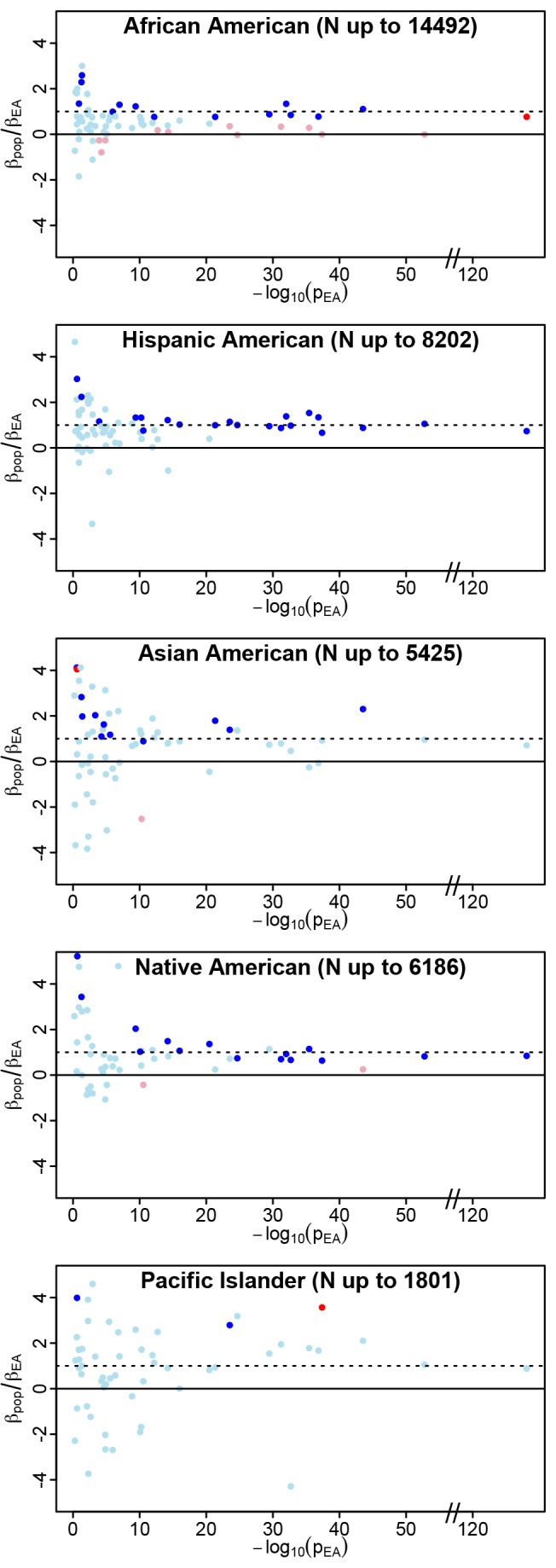
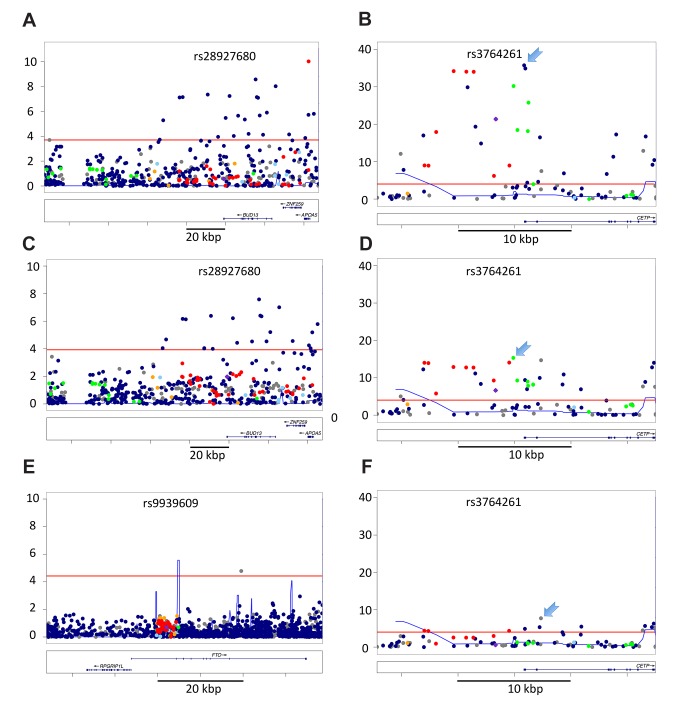
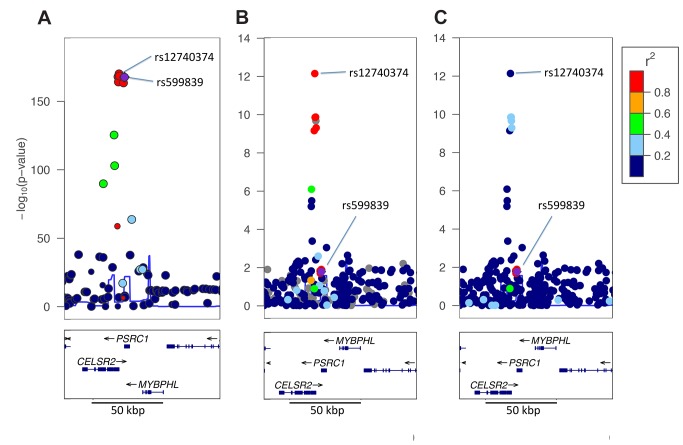
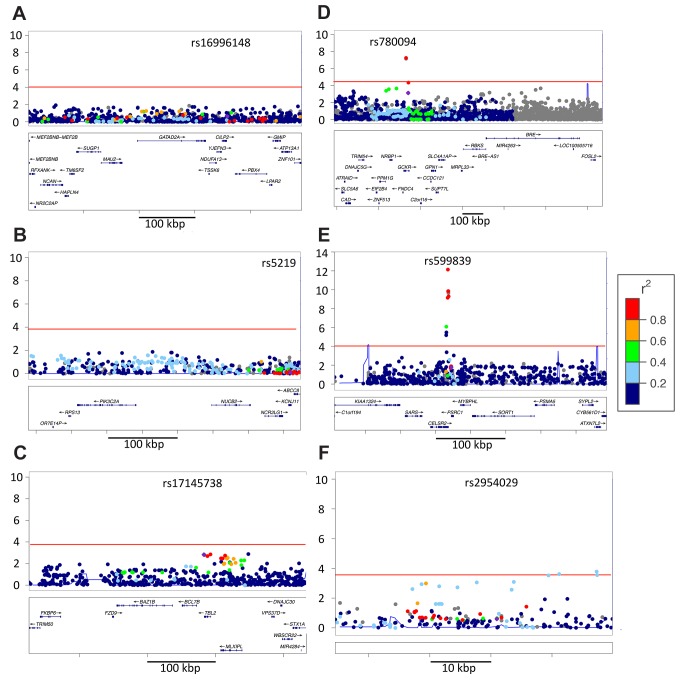
The vast majority of genome-wide association study (GWAS) findings reported to date are from populations with European Ancestry (EA), and it is not yet clear how broadly the genetic associations described will generalize to populations of diverse ancestry. The Population Architecture Using Genomics and Epidemiology (PAGE) study is a consortium of multi-ancestry, population-based studies formed with the objective of refining our understanding of the genetic architecture of common traits emerging from GWAS. In the present analysis of five common diseases and traits, including body mass index, type 2 diabetes, and lipid levels, we compare direction and magnitude of effects for GWAS-identified variants in multiple non-EA populations against EA findings. We demonstrate that, in all populations analyzed, a significant majority of GWAS-identified variants have allelic associations in the same direction as in EA, with none showing a statistically significant effect in the opposite direction, after adjustment for multiple testing. However, 25% of tagSNPs identified in EA GWAS have significantly different effect sizes in at least one non-EA population, and these differential effects were most frequent in African Americans where all differential effects were diluted toward the null. We demonstrate that differential LD between tagSNPs and functional variants within populations contributes significantly to dilute effect sizes in this population. Although most variants identified from GWAS in EA populations generalize to all non-EA populations assessed, genetic models derived from GWAS findings in EA may generate spurious results in non-EA populations due to differential effect sizes. Regardless of the origin of the differential effects, caution should be exercised in applying any genetic risk prediction model based on tagSNPs outside of the ancestry group in which it was derived. Models based directly on functional variation may generalize more robustly, but the identification of functional variants remains challenging.
迄今为止,绝大多数全基因组关联研究 (GWAS) 的发现都来自欧洲血统 (EA) 的人群,目前尚不清楚描述的遗传关联在多大程度上可以推广到具有不同血统的人群。人口基因组学和流行病学研究 (PAGE) 是一个多血统、基于人群的研究联盟,其目标是深化我们对 GWAS 新兴常见特征遗传结构的理解。在对五种常见疾病和特征(包括体重指数、2 型糖尿病和血脂水平)的本分析中,我们比较了多个非 EA 人群中 GWAS 确定的变体与 EA 发现的方向和幅度。我们证明,在所有分析的人群中,GWAS 确定的变体绝大多数具有与 EA 相同的等位基因关联方向,在经过多次测试调整后,没有一个变体显示出统计学上相反方向的显著效应。然而,在至少一个非 EA 人群中,25%的 EA GWAS 中鉴定的标签 SNP 具有显著不同的效应大小,并且这些差异效应在非洲裔美国人中最为频繁,其中所有差异效应都向零值稀释。我们证明,标签 SNP 和人群内功能变体之间的差异 LD 显著导致了该人群中效应大小的稀释。尽管从 EA 人群中的 GWAS 确定的大多数变体可以推广到所有评估的非 EA 人群,但由于效应大小的差异,从 EA GWAS 发现中得出的遗传模型可能会在非 EA 人群中产生虚假结果。无论差异效应的起源如何,在其来源的祖先群体之外,基于标签 SNP 的任何遗传风险预测模型的应用都应谨慎。基于功能变异的模型可能会更稳健地推广,但功能变体的识别仍然具有挑战性。