Department of Obstetrics and Gynecology, Wayne State University, Detroit, MI 48201, USA.
BMC Genomics. 2013 Sep 30;14:666. doi: 10.1186/1471-2164-14-666.
Previous whole-genome shotgun bisulfite sequencing experiments showed that DNA cytosine methylation in the honey bee (Apis mellifera) is almost exclusively at CG dinucleotides in exons. However, the most commonly used method, bisulfite sequencing, cannot distinguish 5-methylcytosine from 5-hydroxymethylcytosine, an oxidized form of 5-methylcytosine that is catalyzed by the TET family of dioxygenases. Furthermore, some analysis software programs under-represent non-CG DNA methylation and hydryoxymethylation for a variety of reasons. Therefore, we used an unbiased analysis of bisulfite sequencing data combined with molecular and bioinformatics approaches to distinguish 5-methylcytosine from 5-hydroxymethylcytosine. By doing this, we have performed the first whole genome analyses of DNA modifications at non-CG sites in honey bees and correlated the effects of these DNA modifications on gene expression and alternative mRNA splicing.
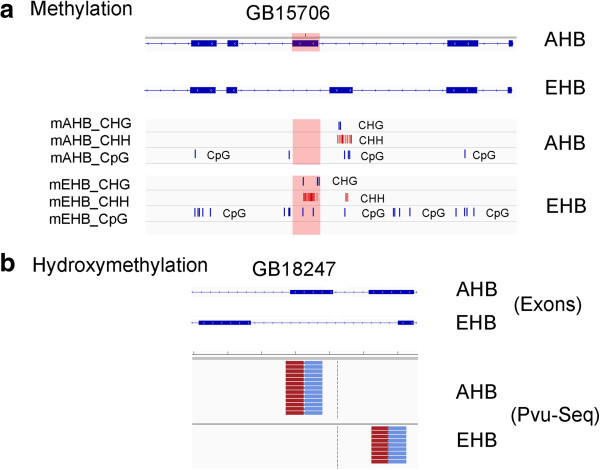
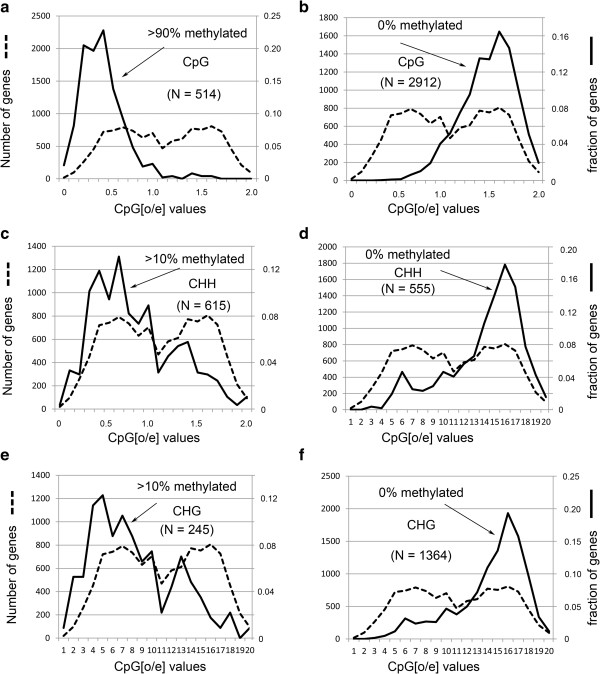
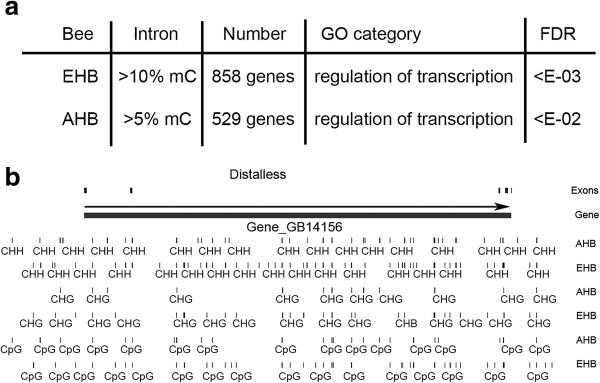
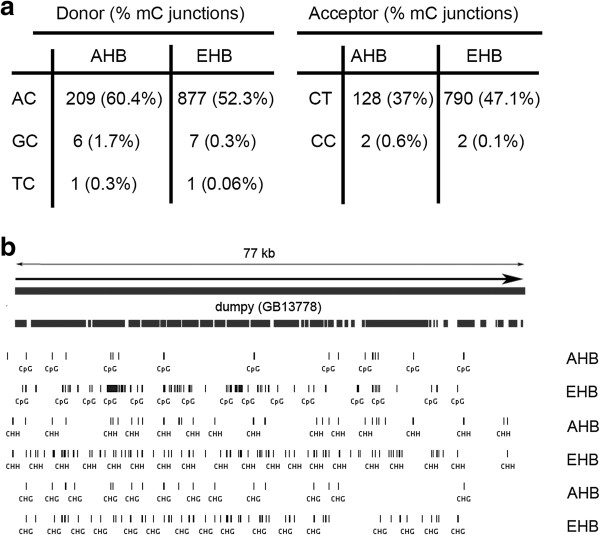
We confirmed, using unbiased analyses of whole-genome shotgun bisulfite sequencing (BS-seq) data, with both new data and published data, the previous finding that CG DNA methylation is enriched in exons in honey bees. However, we also found evidence that cytosine methylation and hydroxymethylation at non-CG sites is enriched in introns. Using antibodies against 5-hydroxmethylcytosine, we confirmed that DNA hydroxymethylation at non-CG sites is enriched in introns. Additionally, using a new technique, Pvu-seq (which employs the enzyme PvuRts1l to digest DNA at 5-hydroxymethylcytosine sites followed by next-generation DNA sequencing), we further confirmed that hydroxymethylation is enriched in introns at non-CG sites.
Cytosine hydroxymethylation at non-CG sites might have more functional significance than previously appreciated, and in honey bees these modifications might be related to the regulation of alternative mRNA splicing by defining the locations of the introns.
先前的全基因组鸟枪法亚硫酸氢盐测序实验表明,在蜜蜂(Apis mellifera)中,DNA 胞嘧啶甲基化几乎完全发生在exon 中的 CG 二核苷酸上。然而,最常用的方法,即亚硫酸氢盐测序,无法区分 5-甲基胞嘧啶和 5-羟甲基胞嘧啶,后者是 5-甲基胞嘧啶的氧化形式,由 TET 家族双氧酶催化产生。此外,由于各种原因,一些分析软件程序会低估非 CG DNA 甲基化和羟甲基化。因此,我们使用了一种无偏倚的方法,对亚硫酸氢盐测序数据进行分析,并结合分子和生物信息学方法,来区分 5-甲基胞嘧啶和 5-羟甲基胞嘧啶。通过这种方法,我们首次对蜜蜂中非 CG 位点的 DNA 修饰进行了全基因组分析,并将这些 DNA 修饰对基因表达和选择性 mRNA 剪接的影响进行了关联。
我们使用无偏倚的全基因组鸟枪法亚硫酸氢盐测序(BS-seq)数据分析,以及新的数据和已发表的数据,证实了先前的发现,即在蜜蜂中 CG DNA 甲基化在 exon 中富集。然而,我们也发现了非 CG 位点的胞嘧啶甲基化和羟甲基化在 intron 中富集的证据。使用针对 5-羟甲基胞嘧啶的抗体,我们证实了非 CG 位点的 DNA 羟甲基化在 intron 中富集。此外,我们使用一种新的技术,Pvu-seq(它使用 PvuRts1l 酶在 5-羟甲基胞嘧啶位点消化 DNA,然后进行下一代 DNA 测序),进一步证实了羟甲基化在非 CG 位点的 intron 中富集。
非 CG 位点的胞嘧啶羟甲基化可能比以前认为的更具有功能意义,在蜜蜂中,这些修饰可能与通过定义 intron 的位置来调节选择性 mRNA 剪接有关。