Klapa Maria I, Tsafou Kalliopi, Theodoridis Evangelos, Tsakalidis Athanasios, Moschonas Nicholas K
Department of General Biology, School of Medicine, University of Patras, Rio, Patras, Greece.
BMC Syst Biol. 2013 Oct 2;7:96. doi: 10.1186/1752-0509-7-96.
Understanding the topology and dynamics of the human protein-protein interaction (PPI) network will significantly contribute to biomedical research, therefore its systematic reconstruction is required. Several meta-databases integrate source PPI datasets, but the protein node sets of their networks vary depending on the PPI data combined. Due to this inherent heterogeneity, the way in which the human PPI network expands via multiple dataset integration has not been comprehensively analyzed. We aim at assembling the human interactome in a global structured way and exploring it to gain insights of biological relevance.
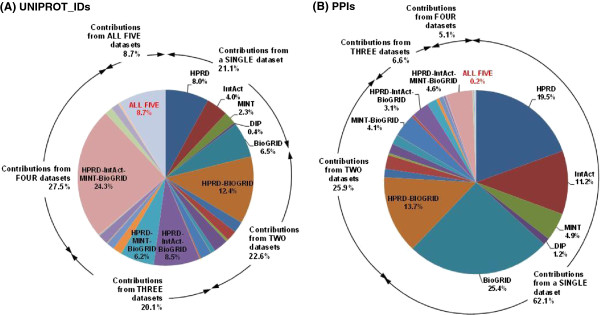
First, we defined the UniProtKB manually reviewed human "complete" proteome as the reference protein-node set and then we mined five major source PPI datasets for direct PPIs exclusively between the reference proteins. We updated the protein and publication identifiers and normalized all PPIs to the UniProt identifier level. The reconstructed interactome covers approximately 60% of the human proteome and has a scale-free structure. No apparent differentiating gene functional classification characteristics were identified for the unrepresented proteins. The source dataset integration augments the network mainly in PPIs. Polyubiquitin emerged as the highest-degree node, but the inclusion of most of its identified PPIs may be reconsidered. The high number (>300) of connections of the subsequent fifteen proteins correlates well with their essential biological role. According to the power-law network structure, the unrepresented proteins should mainly have up to four connections with equally poorly-connected interactors.
Reconstructing the human interactome based on the a priori definition of the protein nodes enabled us to identify the currently included part of the human "complete" proteome, and discuss the role of the proteins within the network topology with respect to their function. As the network expansion has to comply with the scale-free theory, we suggest that the core of the human interactome has essentially emerged. Thus, it could be employed in systems biology and biomedical research, despite the considerable number of currently unrepresented proteins. The latter are probably involved in specialized physiological conditions, justifying the scarcity of related PPI information, and their identification can assist in designing relevant functional experiments and targeted text mining algorithms.
了解人类蛋白质-蛋白质相互作用(PPI)网络的拓扑结构和动力学将极大地促进生物医学研究,因此需要对其进行系统重建。多个元数据库整合了源PPI数据集,但其网络的蛋白质节点集因所合并的PPI数据而异。由于这种固有的异质性,人类PPI网络通过多个数据集整合进行扩展的方式尚未得到全面分析。我们旨在以全局结构化的方式组装人类相互作用组,并对其进行探索以获得生物学相关性的见解。
首先,我们将经过人工审核的UniProtKB人类“完整”蛋白质组定义为参考蛋白质节点集,然后挖掘五个主要的源PPI数据集,以获取仅在参考蛋白质之间的直接PPI。我们更新了蛋白质和出版物标识符,并将所有PPI标准化到UniProt标识符级别。重建的相互作用组覆盖了约60%的人类蛋白质组,具有无标度结构。未被代表的蛋白质未发现明显的差异基因功能分类特征。源数据集整合主要在PPI方面增强了网络。多聚泛素成为度数最高的节点,但对其大多数已鉴定的PPI的纳入可能需要重新考虑。随后的15种蛋白质的大量连接(>300)与其重要的生物学作用密切相关。根据幂律网络结构分析,未被代表的蛋白质与连接性同样较差的相互作用者的连接数应主要不超过4个。
基于蛋白质节点的先验定义重建人类相互作用组,使我们能够识别当前人类“完整”蛋白质组中已包含的部分,并讨论网络拓扑结构中蛋白质相对于其功能的作用。由于网络扩展必须符合无标度理论,我们认为人类相互作用组的核心已基本形成。因此,尽管目前有相当数量的蛋白质未被代表,但它可用于系统生物学和生物医学研究。后者可能参与特殊的生理状况,这解释了相关PPI信息的稀缺性,对它们的识别有助于设计相关的功能实验和有针对性的文本挖掘算法。