Expression Analysis, A Quintiles Company, Durham NC 27713, USA.
BMC Genomics. 2013 Oct 3;14(1):675. doi: 10.1186/1471-2164-14-675.
With the price of next generation sequencing steadily decreasing, bacterial genome assembly is now accessible to a wide range of researchers. It is therefore necessary to understand the best methods for generating a genome assembly, specifically, which combination of sequencing and bioinformatics strategies result in the most accurate assemblies. Here, we sequence three E. coli strains on the Illumina MiSeq, Life Technologies Ion Torrent PGM, and Pacific Biosciences RS. We then perform genome assemblies on all three datasets alone or in combination to determine the best methods for the assembly of bacterial genomes.
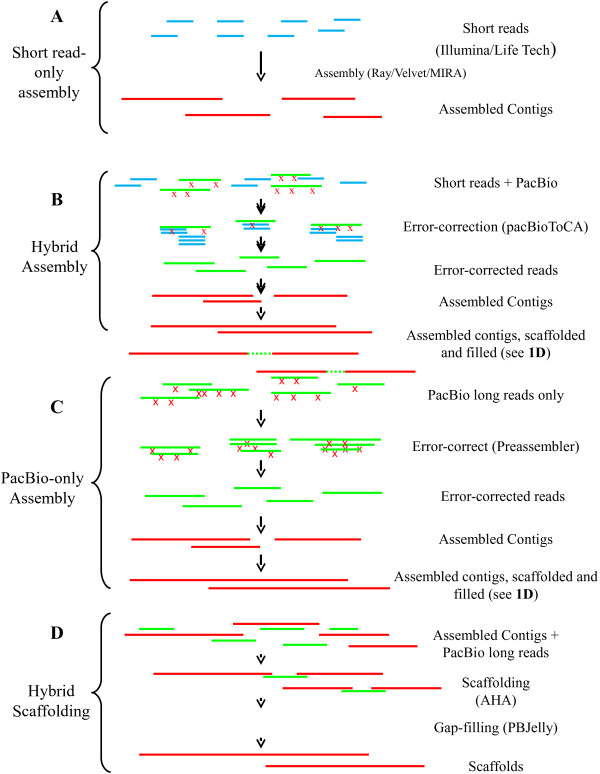
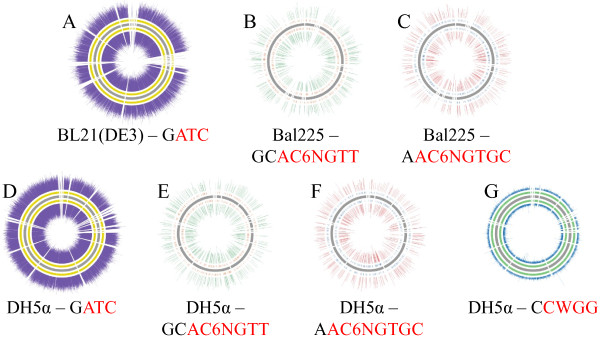
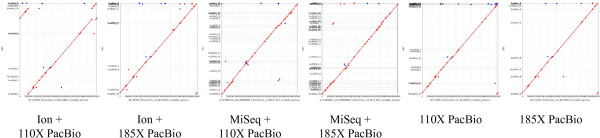
Three E. coli strains - BL21(DE3), Bal225, and DH5α - were sequenced to a depth of 100× on the MiSeq and Ion Torrent machines and to at least 125× on the PacBio RS. Four assembly methods were examined and compared. The previously published BL21(DE3) genome [GenBank:AM946981.2], allowed us to evaluate the accuracy of each of the BL21(DE3) assemblies. BL21(DE3) PacBio-only assemblies resulted in a 90% reduction in contigs versus short read only assemblies, while N50 numbers increased by over 7-fold. Strikingly, the number of SNPs in PacBio-only assemblies were less than half that seen with short read assemblies (20 SNPs vs. ~50 SNPs) and indels also saw dramatic reductions (2 indel >5 bp in PacBio-only assemblies vs. ~12 for short-read only assemblies). Assemblies that used a mixture of PacBio and short read data generally fell in between these two extremes. Use of PacBio sequencing reads also allowed us to call covalent base modifications for the three strains. Each of the strains used here had a known covalent base modification genotype, which was confirmed by PacBio sequencing.
Using data generated solely from the Pacific Biosciences RS, we were able to generate the most complete and accurate de novo assemblies of E. coli strains. We found that the addition of other sequencing technology data offered no improvements over use of PacBio data alone. In addition, the sequencing data from the PacBio RS allowed for sensitive and specific calling of covalent base modifications.
随着下一代测序技术价格的稳步下降,细菌基因组组装现在已经可以为广泛的研究人员所接受。因此,有必要了解生成基因组组装的最佳方法,具体来说,哪种测序和生物信息学策略的组合可以产生最准确的组装。在这里,我们在 Illumina MiSeq、Life Technologies Ion Torrent PGM 和 Pacific Biosciences RS 上对三种大肠杆菌菌株进行测序。然后,我们单独或组合使用所有三个数据集进行基因组组装,以确定细菌基因组组装的最佳方法。
我们对三种大肠杆菌菌株 - BL21(DE3)、Bal225 和 DH5α - 在 MiSeq 和 Ion Torrent 机器上进行了深度为 100×的测序,在 PacBio RS 上至少进行了 125×的测序。我们检查和比较了四种组装方法。之前发表的 BL21(DE3)基因组[GenBank:AM946981.2]使我们能够评估每种 BL21(DE3)组装的准确性。与仅使用短读测序组装相比,BL21(DE3)仅使用 PacBio 测序组装的结果使 contigs 减少了 90%,而 N50 数量增加了 7 倍以上。引人注目的是,PacBio 仅使用测序组装的 SNP 数量不到仅使用短读测序组装的 SNP 数量的一半(20 个 SNP 与50 个 SNP 相比),插入缺失也显著减少(2 个大于 5 bp 的插入缺失在 PacBio 仅使用测序组装中,而短读仅使用测序组装中有12 个)。使用 PacBio 测序reads 和短读数据的混合物的组装通常介于这两个极端之间。使用 PacBio 测序reads 还使我们能够为三种菌株调用共价碱基修饰。这里使用的每种菌株都有一个已知的共价碱基修饰基因型,这是通过 PacBio 测序确认的。
仅使用 Pacific Biosciences RS 生成的数据,我们能够生成最完整和最准确的大肠杆菌菌株从头组装。我们发现,添加其他测序技术数据并没有比仅使用 PacBio 数据带来改进。此外,PacBio RS 的测序数据允许敏感和特异性地调用共价碱基修饰。