Department of Computer Science, Stanford University, Stanford, California 94305, USA;
Genome Res. 2014 Jan;24(1):14-24. doi: 10.1101/gr.155192.113. Epub 2013 Oct 3.
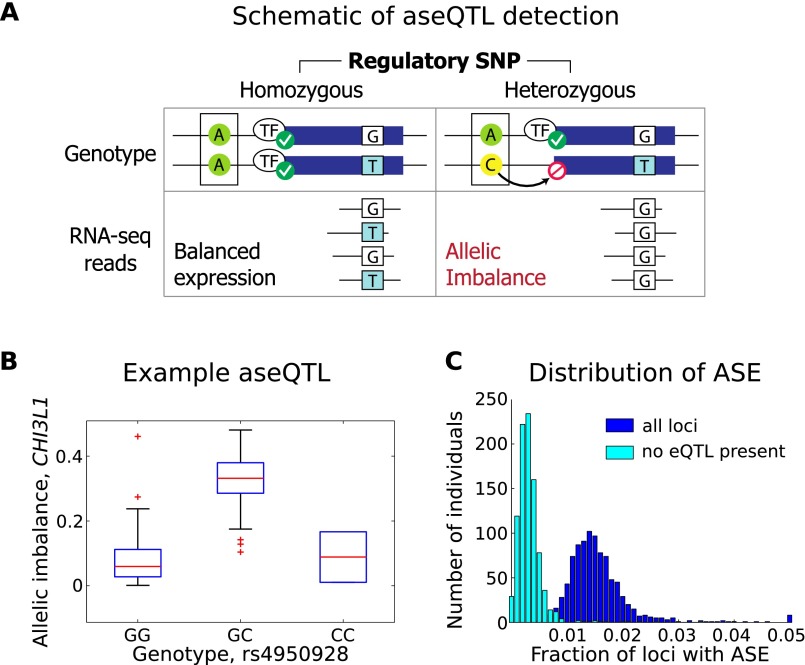
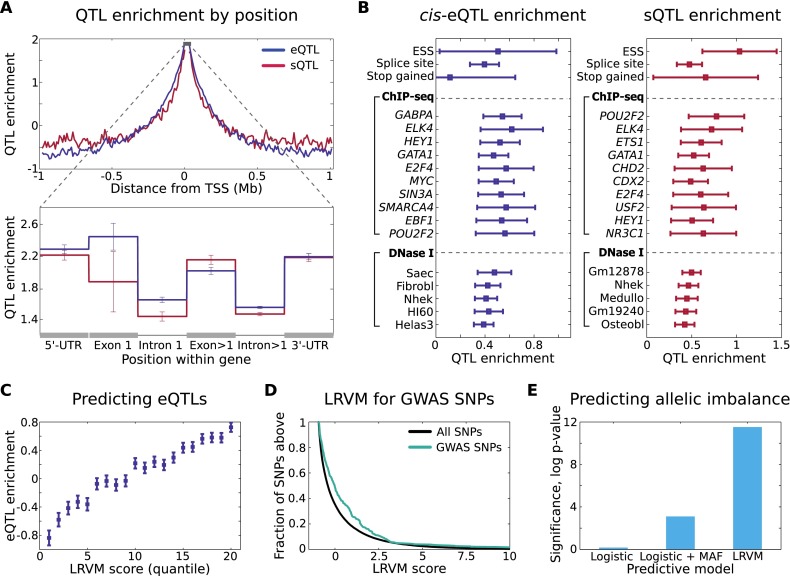
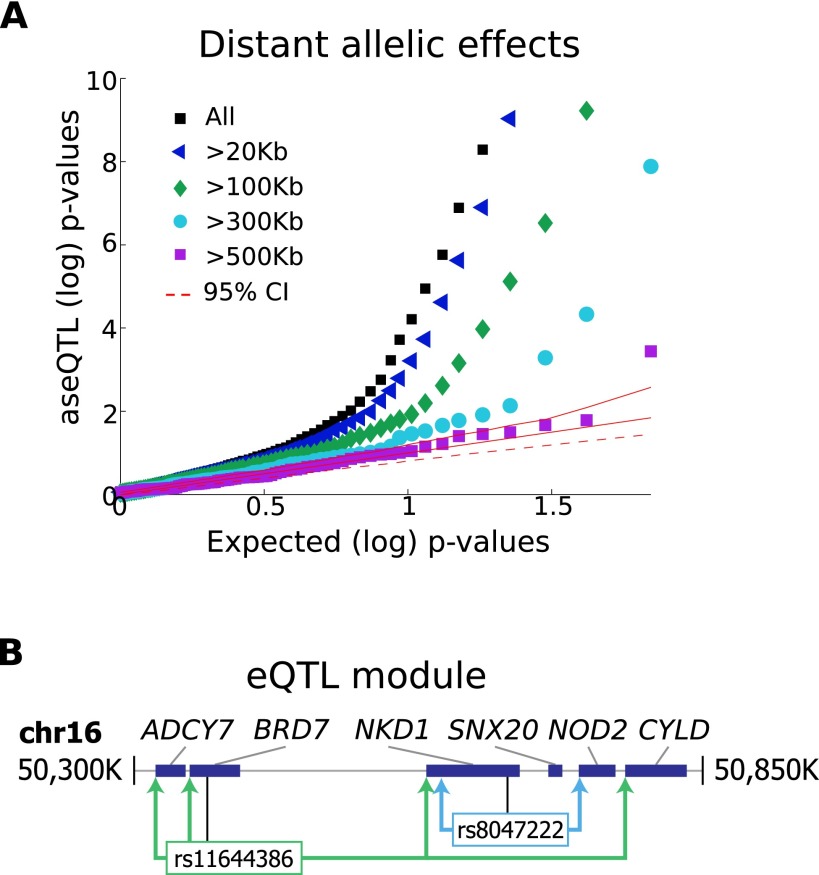
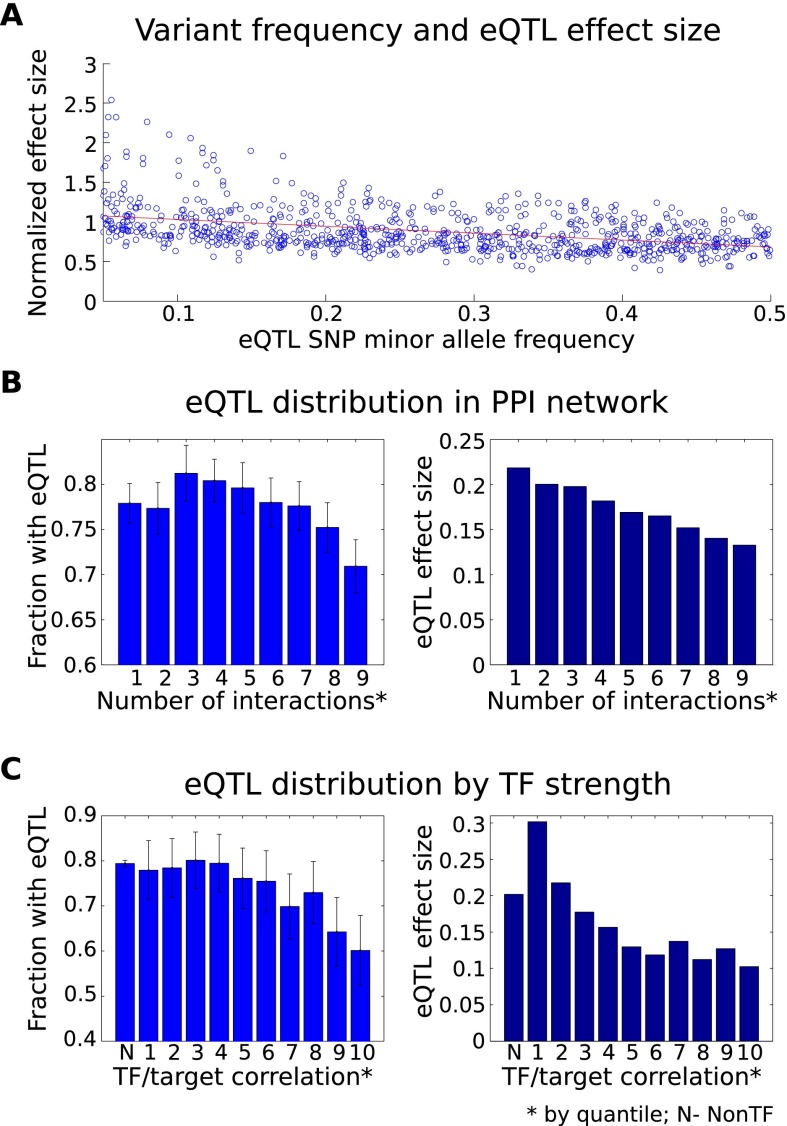
Understanding the consequences of regulatory variation in the human genome remains a major challenge, with important implications for understanding gene regulation and interpreting the many disease-risk variants that fall outside of protein-coding regions. Here, we provide a direct window into the regulatory consequences of genetic variation by sequencing RNA from 922 genotyped individuals. We present a comprehensive description of the distribution of regulatory variation--by the specific expression phenotypes altered, the properties of affected genes, and the genomic characteristics of regulatory variants. We detect variants influencing expression of over ten thousand genes, and through the enhanced resolution offered by RNA-sequencing, for the first time we identify thousands of variants associated with specific phenotypes including splicing and allelic expression. Evaluating the effects of both long-range intra-chromosomal and trans (cross-chromosomal) regulation, we observe modularity in the regulatory network, with three-dimensional chromosomal configuration playing a particular role in regulatory modules within each chromosome. We also observe a significant depletion of regulatory variants affecting central and critical genes, along with a trend of reduced effect sizes as variant frequency increases, providing evidence that purifying selection and buffering have limited the deleterious impact of regulatory variation on the cell. Further, generalizing beyond observed variants, we have analyzed the genomic properties of variants associated with expression and splicing and developed a Bayesian model to predict regulatory consequences of genetic variants, applicable to the interpretation of individual genomes and disease studies. Together, these results represent a critical step toward characterizing the complete landscape of human regulatory variation.
理解人类基因组中调控变异的后果仍然是一个主要挑战,这对于理解基因调控和解释许多位于蛋白质编码区域之外的疾病风险变异具有重要意义。在这里,我们通过对 922 个基因分型个体的 RNA 进行测序,为理解调控变异的后果提供了一个直接的窗口。我们全面描述了调控变异的分布——通过改变的特定表达表型、受影响基因的特性以及调控变异的基因组特征。我们检测到影响一万多个基因表达的变异,并且通过 RNA 测序提供的增强分辨率,我们首次鉴定出数千个与特定表型(包括剪接和等位基因表达)相关的变异。评估长距离染色体内和跨(跨染色体)调控的影响,我们观察到调控网络的模块化,三维染色体构象在每个染色体的调控模块中起着特殊作用。我们还观察到影响中心和关键基因的调控变异明显减少,随着变异频率的增加,效应大小呈下降趋势,这表明净化选择和缓冲作用限制了调控变异对细胞的有害影响。此外,我们超越观察到的变异进行推广,分析了与表达和剪接相关的变异的基因组特性,并开发了一种贝叶斯模型来预测遗传变异的调控后果,适用于个体基因组和疾病研究的解释。总之,这些结果代表了朝着描述人类调控变异全貌迈出的关键一步。