Department of Agricultural, Food, and Environmental Sciences, University of Pisa, Via del Borghetto 80, I-56124 Pisa, Italy.
BMC Genomics. 2013 Oct 6;14:686. doi: 10.1186/1471-2164-14-686.
Next generation sequencing provides a powerful tool to study genome structure in species whose genomes are far from being completely sequenced. In this work we describe and compare different computational approaches to evaluate the repetitive component of the genome of sunflower, by using medium/low coverage Illumina or 454 libraries.
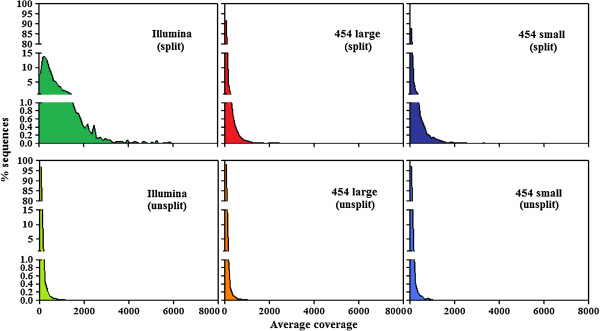
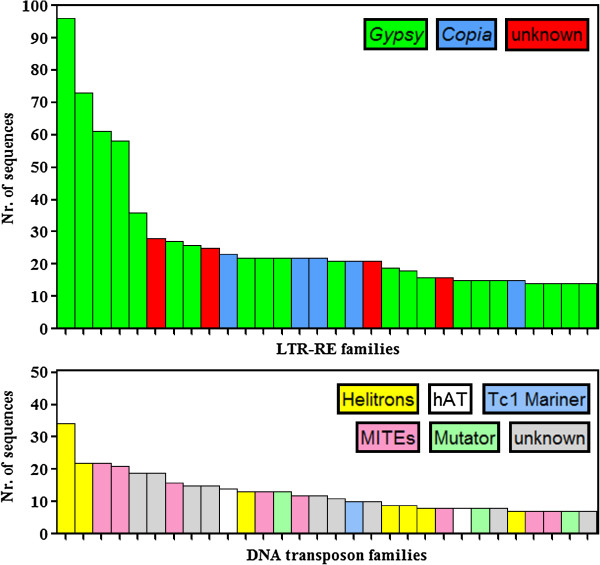
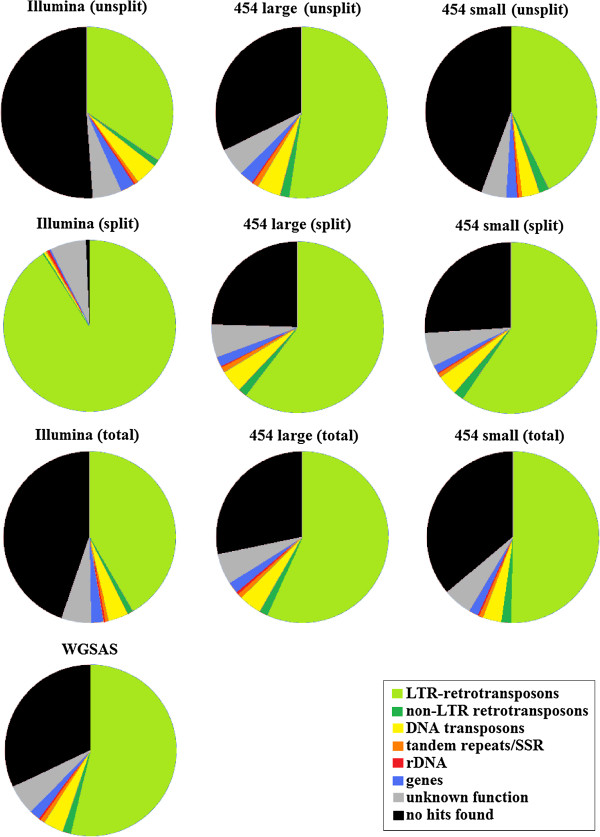
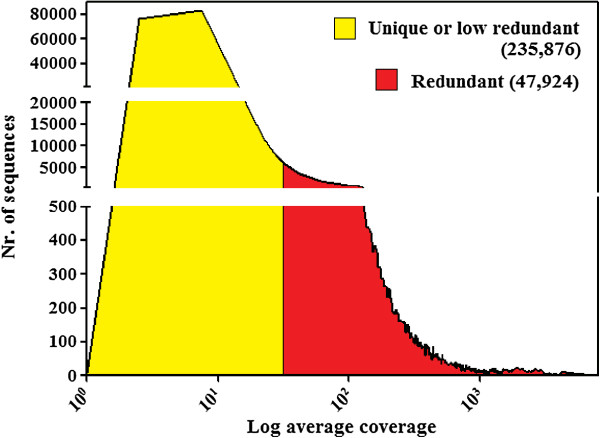
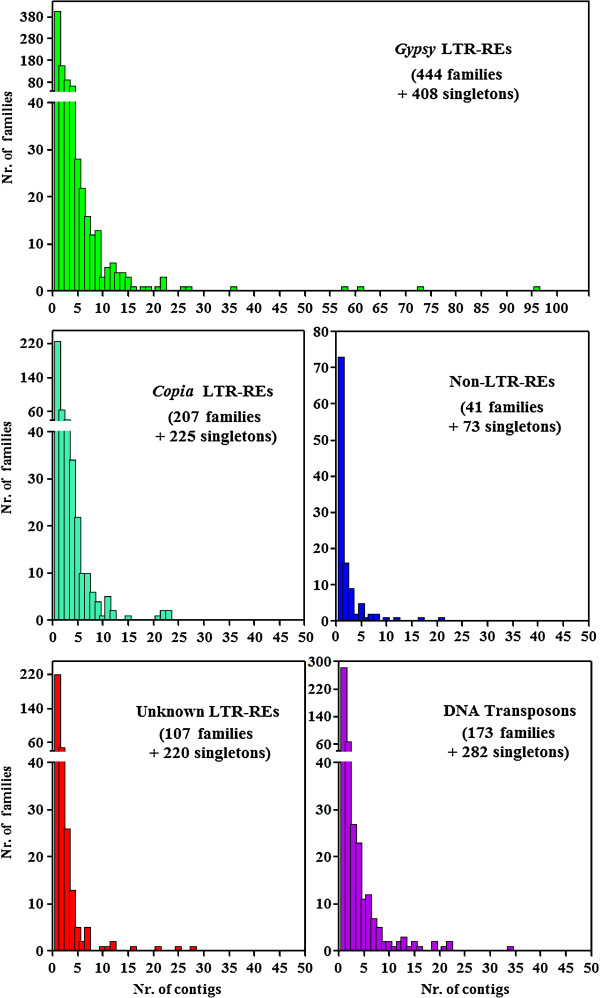
By varying sequencing technology (Illumina or 454), coverage (0.55 x-1.25 x), assemblers and assembly procedures, six different genomic databases were produced. The annotation of these databases showed that they were composed of different proportions of repetitive DNA families. The final assembly of the sequences belonging to the six databases produced a whole genome set of 283,800 contigs. The redundancy of each contig was estimated by mapping the whole genome set with a large Illumina read set and measuring the number of matched Illumina reads. The repetitive component amounted to 81% of the sunflower genome, that is composed mainly of numerous families of Gypsy and Copia retrotransposons. Also many families of non autonomous retrotransposons and DNA transposons (especially of the Helitron superfamily) were identified.
The results substantially matched those previously obtained by using a Sanger-sequenced shotgun library and a standard 454 whole-genome-shotgun approach, indicating the reliability of the proposed procedures also for other species. The repetitive sequences were collected to produce a database, SUNREP, that will be useful for the annotation of the sunflower genome sequence and for studying the genome evolution in dicotyledons.
下一代测序为研究基因组结构提供了强大的工具,这些基因组远未完全测序。在这项工作中,我们描述并比较了不同的计算方法,以评估利用中/低覆盖度的 Illumina 或 454 文库对向日葵基因组重复成分的评估。
通过改变测序技术(Illumina 或 454)、覆盖度(0.55x-1.25x)、组装器和组装程序,产生了六个不同的基因组数据库。这些数据库的注释表明,它们由不同比例的重复 DNA 家族组成。属于六个数据库的序列的最终组装产生了一个 283800 个序列的全基因组集。通过用大量的 Illumina 读取集对整个基因组集进行映射,并测量匹配的 Illumina 读取的数量,估计了每个序列的冗余性。重复成分占向日葵基因组的 81%,主要由大量的 Gypsy 和 Copia 反转录转座子家族组成。还鉴定了许多非自主反转录转座子和 DNA 转座子(特别是 Helitron 超家族)家族。
结果与以前使用 Sanger 测序的鸟枪法文库和标准的 454 全基因组鸟枪法获得的结果基本一致,表明所提出的方法对于其他物种也是可靠的。重复序列被收集起来以产生一个数据库,SUNREP,这将有助于向日葵基因组序列的注释和研究双子叶植物的基因组进化。