Duque Thyago, Samee Md Abul Hassan, Kazemian Majid, Pham Hannah N, Brodsky Michael H, Sinha Saurabh
Department of Computer Science, University of Illinois at Urbana-Champaign.
Mol Biol Evol. 2014 Jan;31(1):184-200. doi: 10.1093/molbev/mst170. Epub 2013 Oct 4.
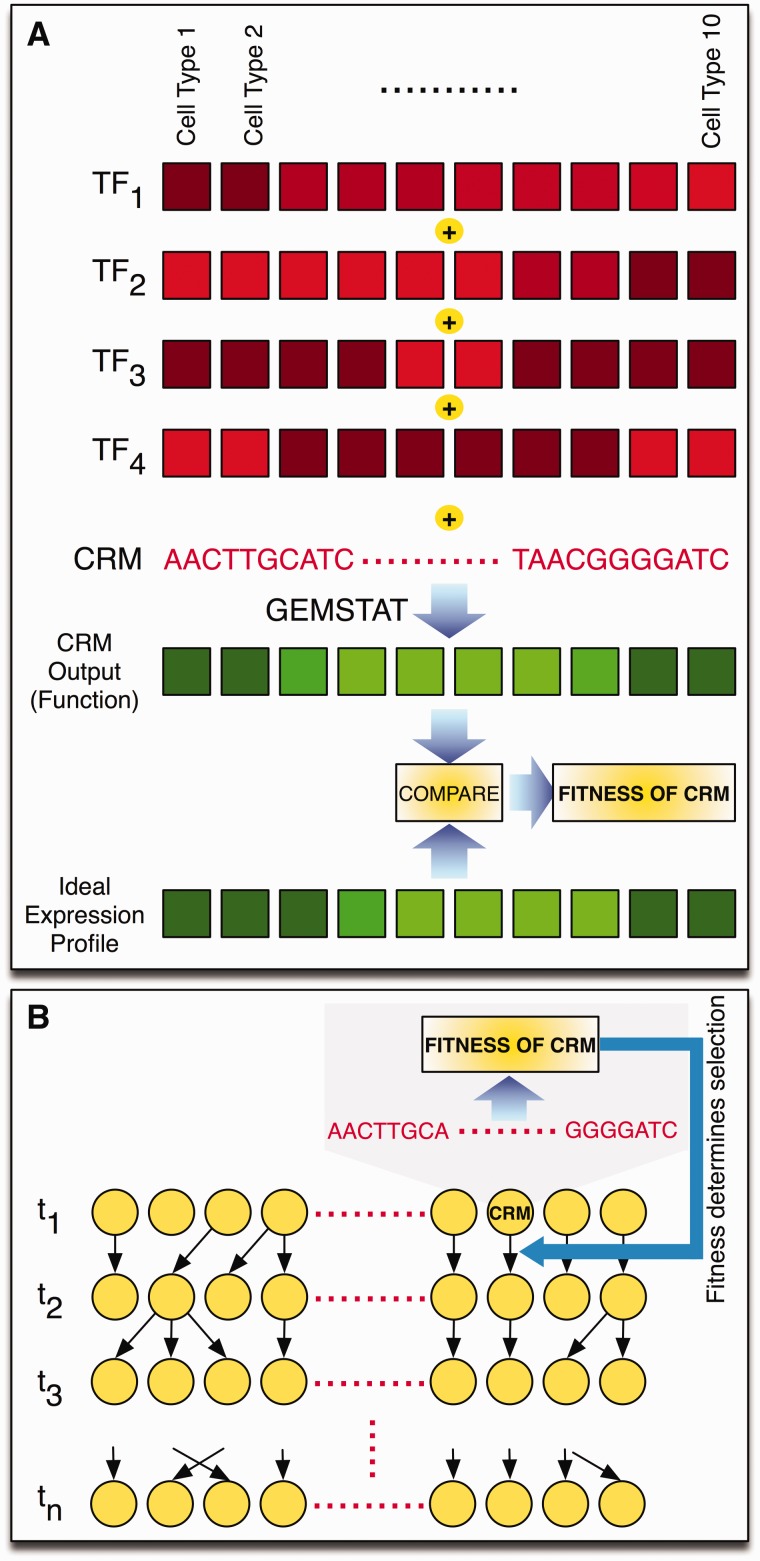
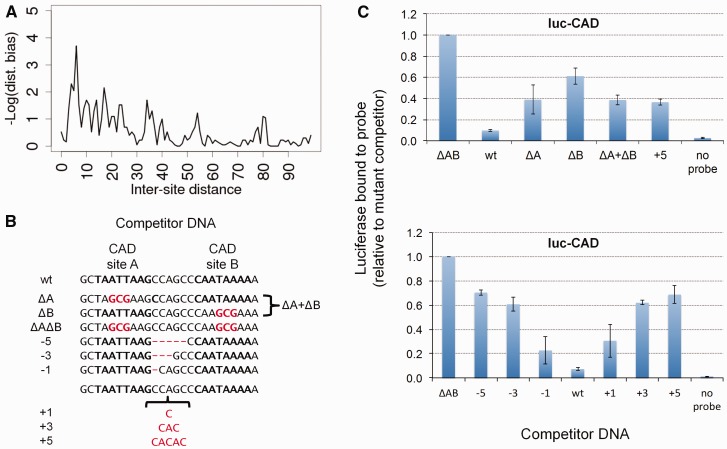
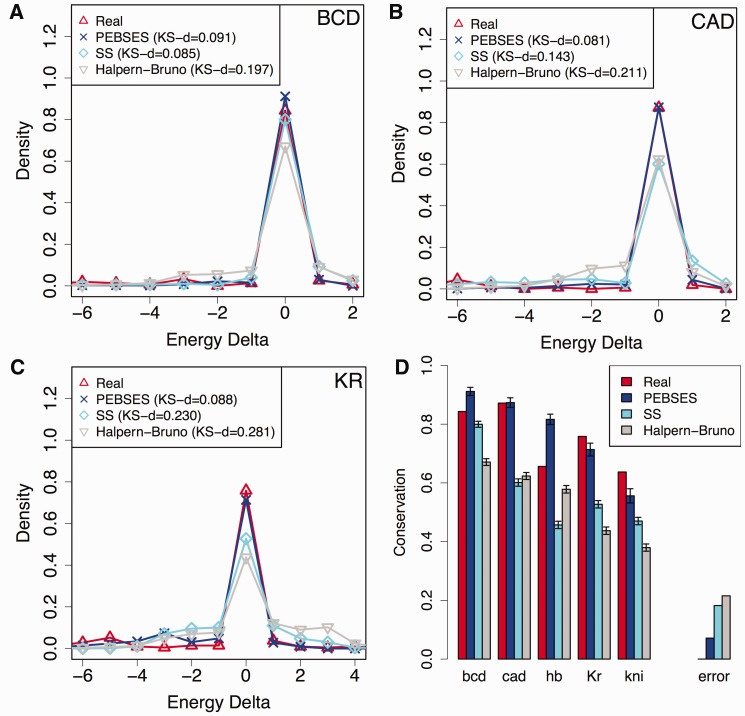
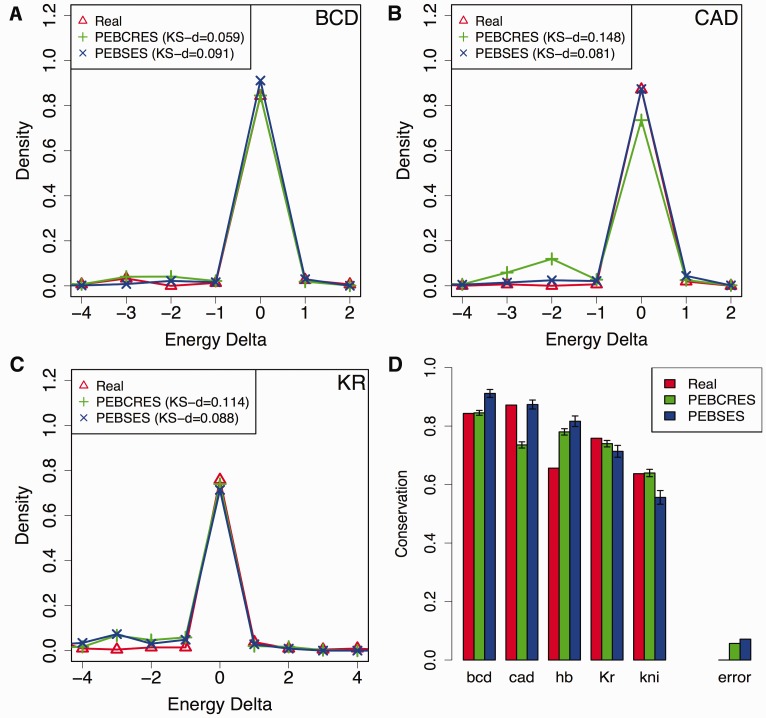
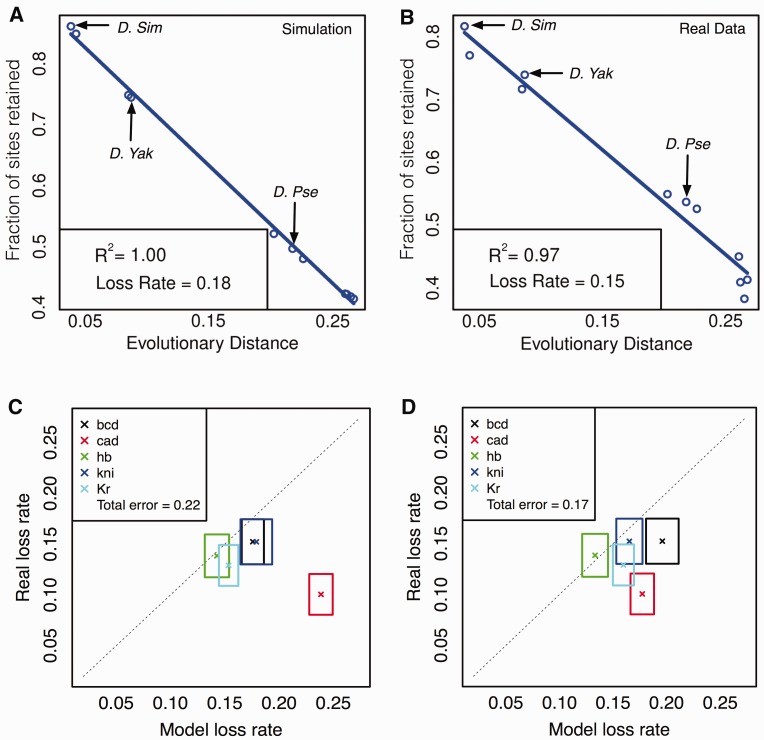
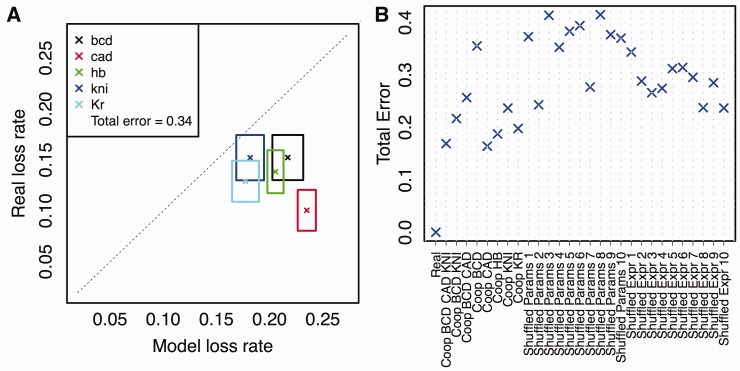
There is growing interest in models of regulatory sequence evolution. However, existing models specifically designed for regulatory sequences consider the independent evolution of individual transcription factor (TF)-binding sites, ignoring that the function and evolution of a binding site depends on its context, typically the cis-regulatory module (CRM) in which the site is located. Moreover, existing models do not account for the gene-specific roles of TF-binding sites, primarily because their roles often are not well understood. We introduce two models of regulatory sequence evolution that address some of the shortcomings of existing models and implement simulation frameworks based on them. One model simulates the evolution of an individual binding site in the context of a CRM, while the other evolves an entire CRM. Both models use a state-of-the art sequence-to-expression model to predict the effects of mutations on the regulatory output of the CRM and determine the strength of selection. We use the new framework to simulate the evolution of TF-binding sites in 37 well-studied CRMs belonging to the anterior-posterior patterning system in Drosophila embryos. We show that these simulations provide accurate fits to evolutionary data from 12 Drosophila genomes, which includes statistics of binding site conservation on relatively short evolutionary scales and site loss across larger divergence times. The new framework allows us, for the first time, to test hypotheses regarding the underlying cis-regulatory code by directly comparing the evolutionary implications of the hypothesis with the observed evolutionary dynamics of binding sites. Using this capability, we find that explicitly modeling self-cooperative DNA binding by the TF Caudal (CAD) provides significantly better fits than an otherwise identical evolutionary simulation that lacks this mechanistic aspect. This hypothesis is further supported by a statistical analysis of the distribution of intersite spacing between adjacent CAD sites. Experimental tests confirm direct homodimeric interaction between CAD molecules as well as self-cooperative DNA binding by CAD. We note that computational modeling of the D. melanogaster CRMs alone did not yield significant evidence to support CAD self-cooperativity. We thus demonstrate how specific mechanistic details encoded in CRMs can be revealed by modeling their evolution and fitting such models to multispecies data.
人们对调控序列进化模型的兴趣与日俱增。然而,现有的专门针对调控序列设计的模型考虑的是单个转录因子(TF)结合位点的独立进化,却忽略了结合位点的功能和进化取决于其所处的环境,通常是该位点所在的顺式调控模块(CRM)。此外,现有模型没有考虑TF结合位点的基因特异性作用,主要原因是其作用往往尚未得到充分理解。我们引入了两种调控序列进化模型,解决了现有模型的一些不足之处,并基于它们实现了模拟框架。一种模型模拟CRM环境中单个结合位点的进化,而另一种则使整个CRM进化。两种模型都使用了先进的序列到表达模型来预测突变对CRM调控输出的影响,并确定选择强度。我们使用新框架来模拟果蝇胚胎前后模式形成系统中37个经过充分研究的CRM中TF结合位点的进化。我们表明,这些模拟与来自12个果蝇基因组的进化数据精确拟合,这些数据包括相对较短进化尺度上结合位点保守性的统计以及更大分歧时间内位点丢失的情况。新框架首次使我们能够通过直接比较假设的进化含义与观察到的结合位点进化动态来检验关于潜在顺式调控密码的假设。利用这一能力,我们发现通过TF尾(CAD)对自协同DNA结合进行明确建模,比缺乏这一机制方面的相同进化模拟能提供显著更好的拟合。相邻CAD位点之间位点间距分布的统计分析进一步支持了这一假设。实验测试证实了CAD分子之间的直接同二聚体相互作用以及CAD的自协同DNA结合。我们注意到,仅对黑腹果蝇CRM进行计算建模并没有产生支持CAD自协同性的重要证据。因此,我们展示了如何通过对CRM的进化进行建模并将此类模型与多物种数据拟合,来揭示CRM中编码的特定机制细节。