Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, IL, USA.
PLoS Genet. 2013;9(8):e1003571. doi: 10.1371/journal.pgen.1003571. Epub 2013 Aug 1.
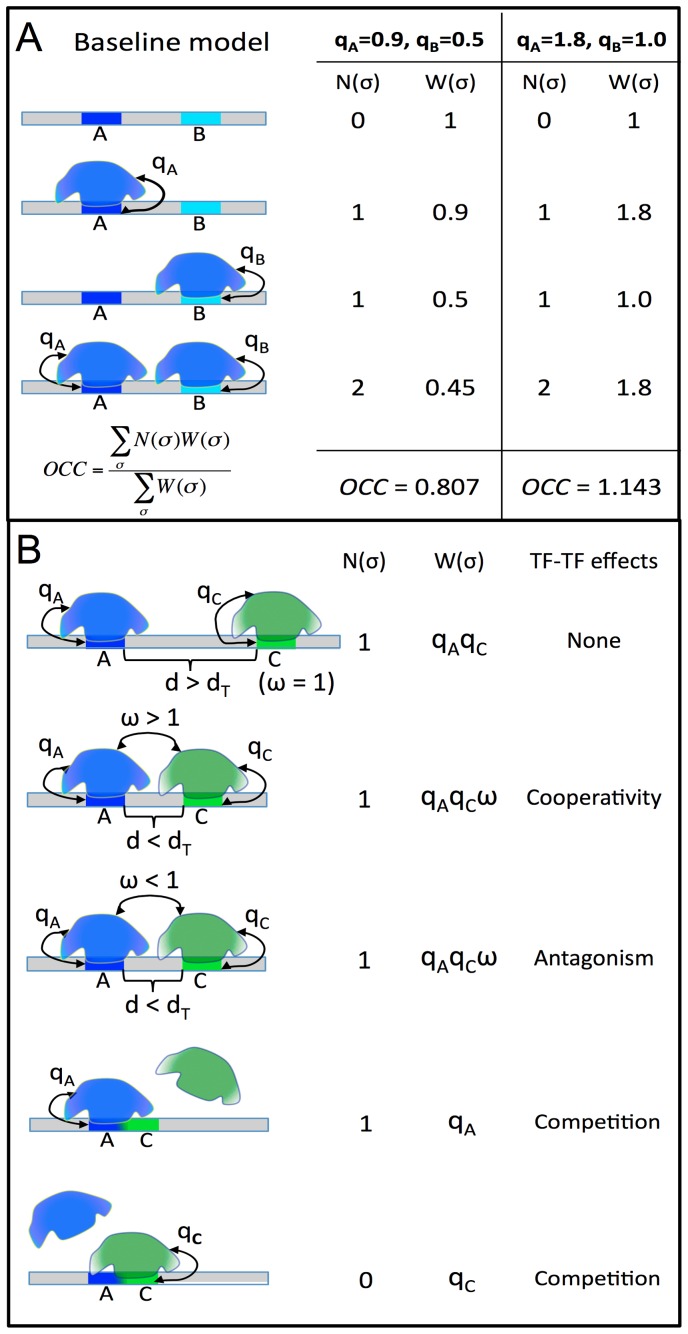
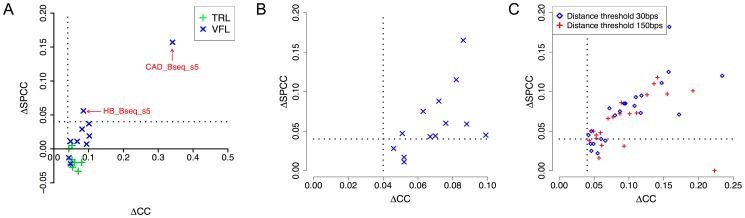
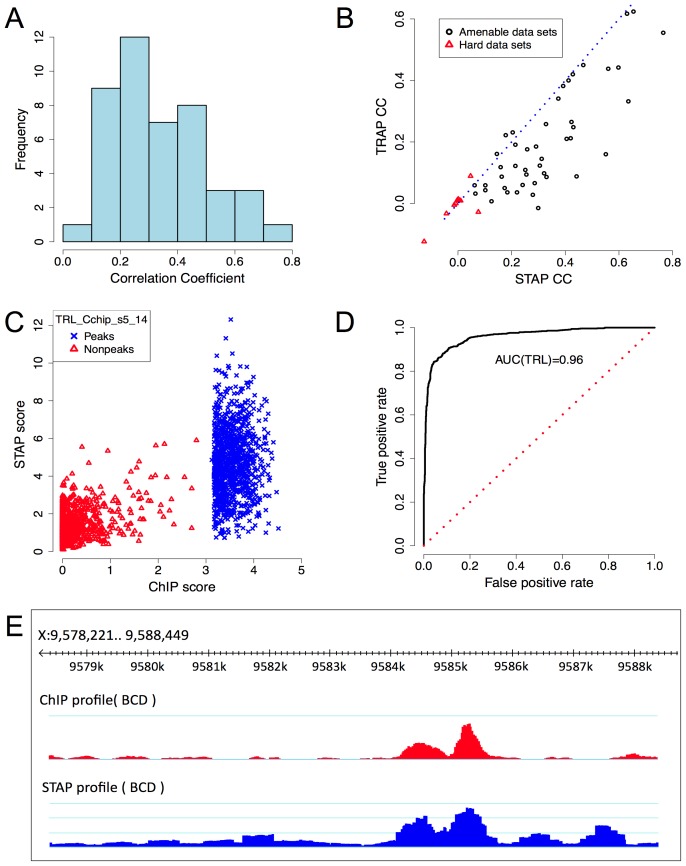
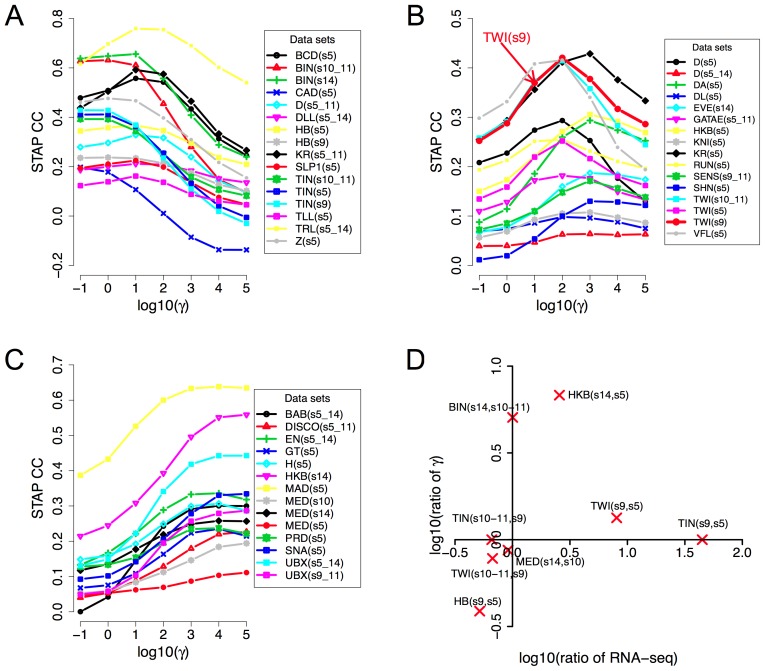
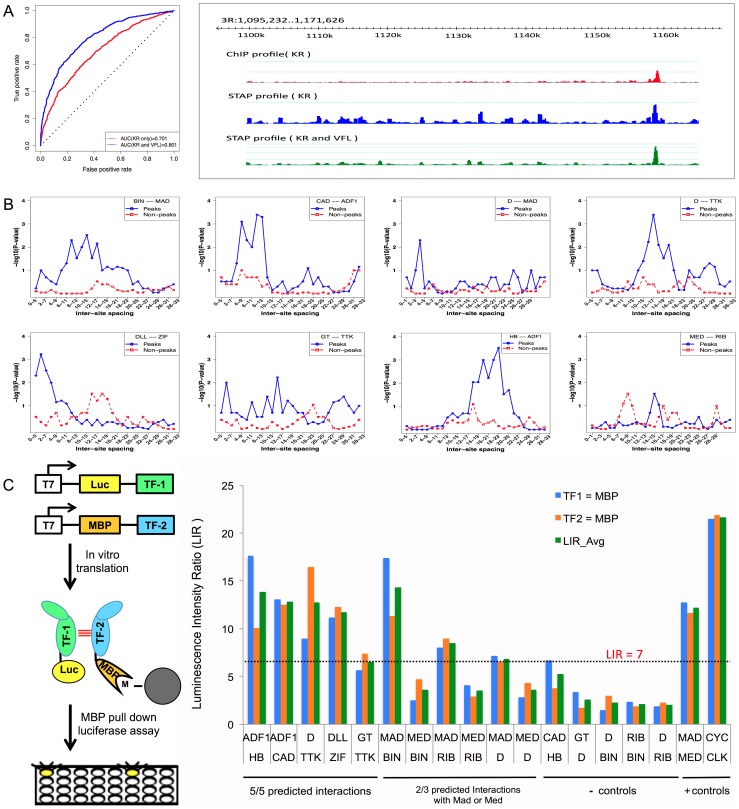
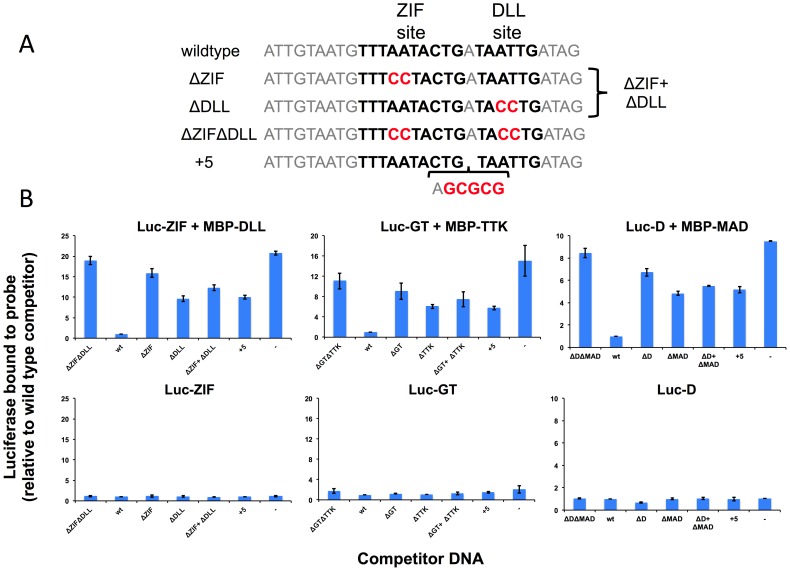
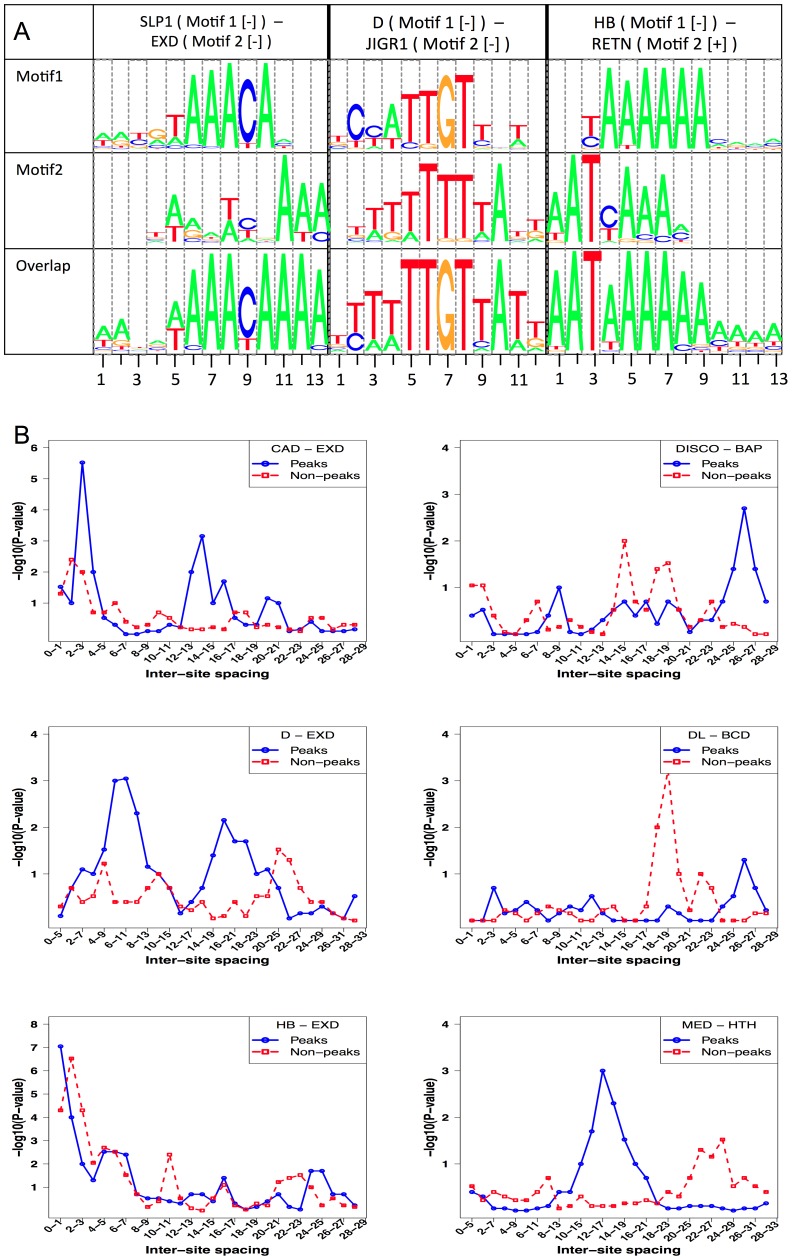
ChIP-based genome-wide assays of transcription factor (TF) occupancy have emerged as a powerful, high-throughput method to understand transcriptional regulation, especially on a global scale. This has led to great interest in the underlying biochemical mechanisms that direct TF-DNA binding, with the ultimate goal of computationally predicting a TF's occupancy profile in any cellular condition. In this study, we examined the influence of various potential determinants of TF-DNA binding on a much larger scale than previously undertaken. We used a thermodynamics-based model of TF-DNA binding, called "STAP," to analyze 45 TF-ChIP data sets from Drosophila embryonic development. We built a cross-validation framework that compares a baseline model, based on the ChIP'ed ("primary") TF's motif, to more complex models where binding by secondary TFs is hypothesized to influence the primary TF's occupancy. Candidates interacting TFs were chosen based on RNA-SEQ expression data from the time point of the ChIP experiment. We found widespread evidence of both cooperative and antagonistic effects by secondary TFs, and explicitly quantified these effects. We were able to identify multiple classes of interactions, including (1) long-range interactions between primary and secondary motifs (separated by ≤150 bp), suggestive of indirect effects such as chromatin remodeling, (2) short-range interactions with specific inter-site spacing biases, suggestive of direct physical interactions, and (3) overlapping binding sites suggesting competitive binding. Furthermore, by factoring out the previously reported strong correlation between TF occupancy and DNA accessibility, we were able to categorize the effects into those that are likely to be mediated by the secondary TF's effect on local accessibility and those that utilize accessibility-independent mechanisms. Finally, we conducted in vitro pull-down assays to test model-based predictions of short-range cooperative interactions, and found that seven of the eight TF pairs tested physically interact and that some of these interactions mediate cooperative binding to DNA.
基于 ChIP 的转录因子(TF)占有率全基因组分析已成为一种强大的高通量方法,可用于了解转录调控,尤其是在全局范围内。这引发了人们对指导 TF-DNA 结合的潜在生化机制的极大兴趣,其最终目标是在任何细胞状态下计算预测 TF 的占有率。在这项研究中,我们以比以往更大的规模研究了各种可能决定 TF-DNA 结合的因素的影响。我们使用了一种基于热力学的 TF-DNA 结合模型,称为“STAP”,来分析来自果蝇胚胎发育的 45 个 TF-ChIP 数据集。我们构建了一个交叉验证框架,将基于 ChIP'ed(“主要”)TF 基序的基线模型与更复杂的模型进行比较,在这些模型中,假设次要 TF 的结合会影响主要 TF 的占有率。候选相互作用的 TF 是根据 ChIP 实验时间点的 RNA-SEQ 表达数据选择的。我们发现次要 TF 具有广泛的协同和拮抗作用的证据,并明确量化了这些作用。我们能够识别出多种类型的相互作用,包括:(1)主要和次要基序之间的长程相互作用(间隔 ≤150bp),提示间接作用,如染色质重塑;(2)具有特定的局域间隔偏好的短程相互作用,提示直接的物理相互作用;(3)重叠的结合位点提示竞争结合。此外,通过消除以前报道的 TF 占有率与 DNA 可及性之间的强相关性,我们能够将这些作用分为可能是由次要 TF 对局部可及性的影响介导的作用,以及利用独立于可及性的机制的作用。最后,我们进行了体外下拉测定以测试基于模型的短程协同作用预测,发现测试的八个 TF 对中的七个具有物理相互作用,并且这些相互作用中的一些介导了对 DNA 的协同结合。