Institute of Information Science, Academia Sinica, Taipei, Taiwan.
BMC Bioinformatics. 2013 Oct 11;14:304. doi: 10.1186/1471-2105-14-304.
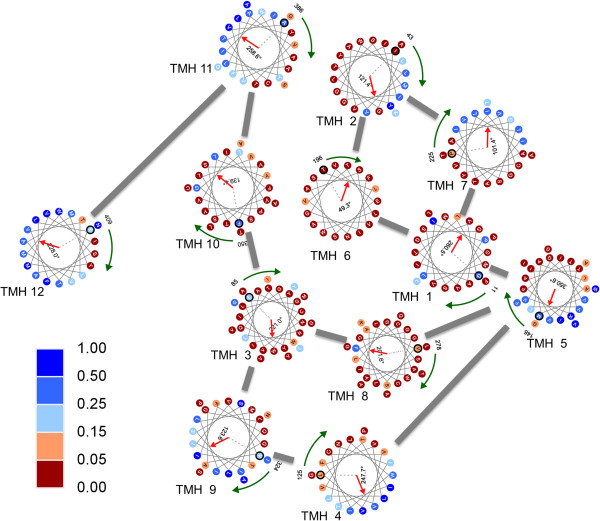
Since membrane protein structures are challenging to crystallize, computational approaches are essential for elucidating the sequence-to-structure relationships. Structural modeling of membrane proteins requires a multidimensional approach, and one critical geometric parameter is the rotational angle of transmembrane helices. Rotational angles of transmembrane helices are characterized by their folded structures and could be inferred by the hydrophobic moment; however, the folding mechanism of membrane proteins is not yet fully understood. The rotational angle of a transmembrane helix is related to the exposed surface of a transmembrane helix, since lipid exposure gives the degree of accessibility of each residue in lipid environment. To the best of our knowledge, there have been few advances in investigating whether an environment descriptor of lipid exposure could infer a geometric parameter of rotational angle.
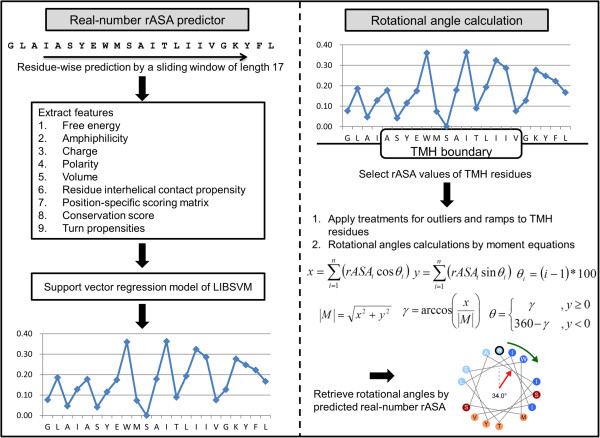
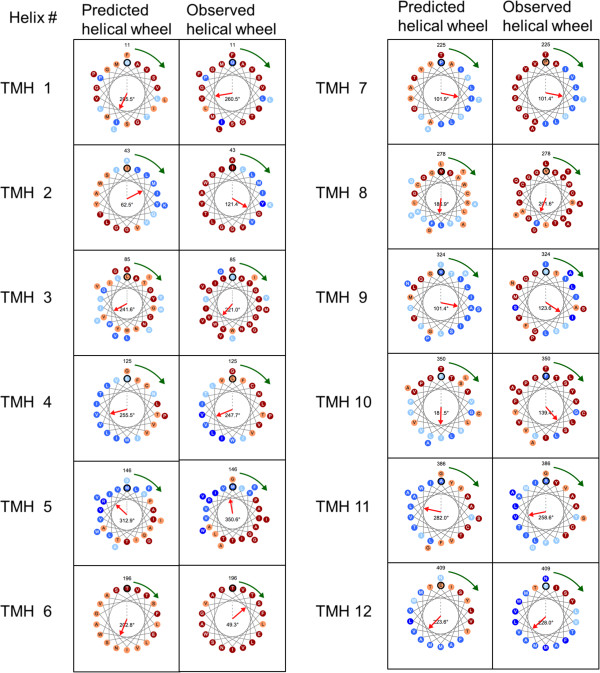
Here, we present an analysis of the relationship between rotational angles and lipid exposure and a support-vector-machine method, called TMexpo, for predicting both structural features from sequences. First, we observed from the development set of 89 protein chains that the lipid exposure, i.e., the relative accessible surface area (rASA) of residues in the lipid environment, generated from high-resolution protein structures could infer the rotational angles with a mean absolute angular error (MAAE) of 46.32˚. More importantly, the predicted rASA from TMexpo achieved an MAAE of 51.05˚, which is better than 71.47˚ obtained by the best of the compared hydrophobicity scales. Lastly, TMexpo outperformed the compared methods in rASA prediction on the independent test set of 21 protein chains and achieved an overall Matthew's correlation coefficient, accuracy, sensitivity, specificity, and precision of 0.51, 75.26%, 81.30%, 69.15%, and 72.73%, respectively. TMexpo is publicly available at http://bio-cluster.iis.sinica.edu.tw/TMexpo.
TMexpo can better predict rASA and rotational angles than the compared methods. When rotational angles can be accurately predicted, free modeling of transmembrane protein structures in turn may benefit from a reduced complexity in ensembles with a significantly less number of packing arrangements. Furthermore, sequence-based prediction of both rotational angle and lipid exposure can provide essential information when high-resolution structures are unavailable and contribute to experimental design to elucidate transmembrane protein functions.
由于膜蛋白结构难以结晶,因此计算方法对于阐明序列-结构关系至关重要。膜蛋白的结构建模需要多维方法,一个关键的几何参数是跨膜螺旋的旋转角度。跨膜螺旋的旋转角度由其折叠结构决定,可以通过疏水力矩推断出来;然而,膜蛋白的折叠机制尚未完全理解。跨膜螺旋的旋转角度与跨膜螺旋的暴露表面有关,因为脂质暴露程度给出了每个残基在脂质环境中的可及度。据我们所知,关于环境描述符的脂质暴露是否可以推断出旋转角度的几何参数,这方面的研究进展甚少。
在这里,我们分析了旋转角度与脂质暴露之间的关系,并提出了一种支持向量机方法 TMexpo,用于从序列预测结构特征。首先,我们从 89 条蛋白质链的开发集观察到,来自高分辨率蛋白质结构的脂质暴露,即残基在脂质环境中的相对可及表面积 (rASA),可以推断出旋转角度,平均绝对角度误差 (MAAE) 为 46.32˚。更重要的是,TMexpo 预测的 rASA 的平均绝对角度误差为 51.05˚,优于比较的疏水性尺度中最好的 71.47˚。最后,TMexpo 在 21 条蛋白质链的独立测试集上的 rASA 预测中优于比较方法,总体马修相关系数、准确性、敏感性、特异性和精确性分别为 0.51、75.26%、81.30%、69.15%和 72.73%。TMexpo 可在 http://bio-cluster.iis.sinica.edu.tw/TMexpo 上公开获取。
TMexpo 可以比比较方法更好地预测 rASA 和旋转角度。当旋转角度可以准确预测时,跨膜蛋白质结构的自由建模反过来可能会受益于具有显著较少包装排列的集合的复杂性降低。此外,当无法获得高分辨率结构时,基于序列的旋转角度和脂质暴露预测可以提供必要的信息,并有助于阐明跨膜蛋白质功能的实验设计。