Bioinformatics Research Group, Bina Nusantara University, Jakarta, Indonesia.
BMC Bioinformatics. 2013 Oct 23;14:312. doi: 10.1186/1471-2105-14-312.
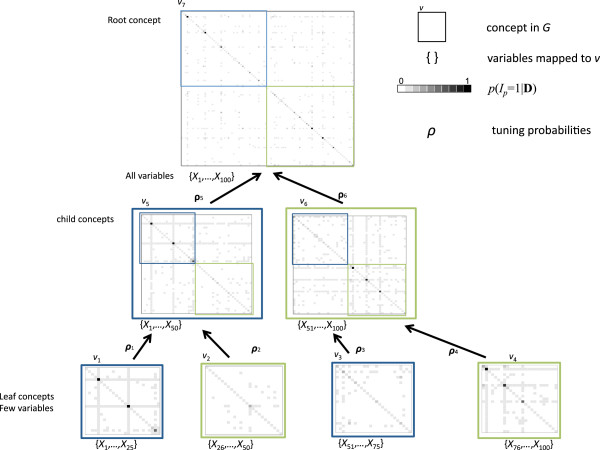
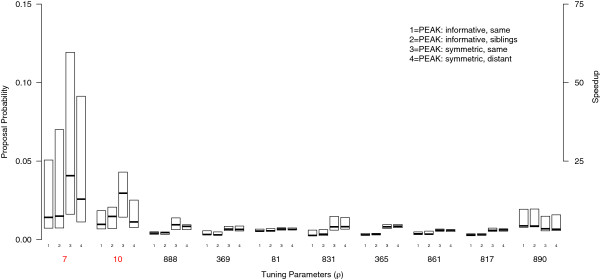
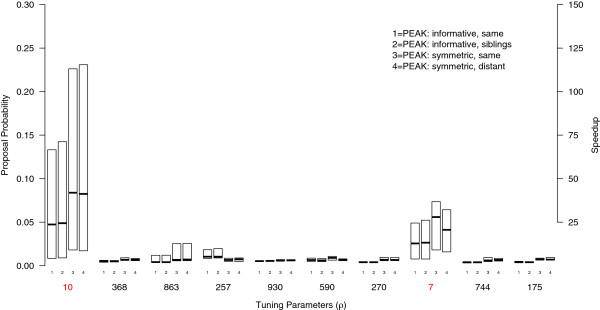
Testing for marginal associations between numerous genetic variants and disease may miss complex relationships among variables (e.g., gene-gene interactions). Bayesian approaches can model multiple variables together and offer advantages over conventional model building strategies, including using existing biological evidence as modeling priors and acknowledging that many models may fit the data well. With many candidate variables, Bayesian approaches to variable selection rely on algorithms to approximate the posterior distribution of models, such as Markov-Chain Monte Carlo (MCMC). Unfortunately, MCMC is difficult to parallelize and requires many iterations to adequately sample the posterior. We introduce a scalable algorithm called PEAK that improves the efficiency of MCMC by dividing a large set of variables into related groups using a rooted graph that resembles a mountain peak. Our algorithm takes advantage of parallel computing and existing biological databases when available.
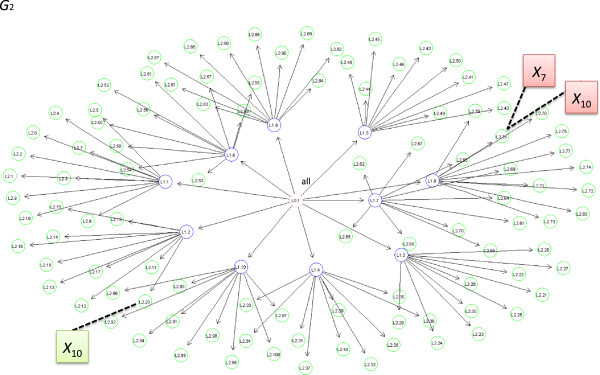
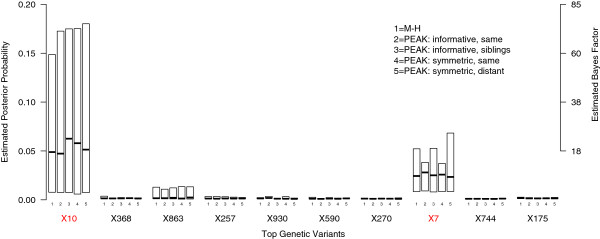
By using graphs to manage a model space with more than 500,000 candidate variables, we were able to improve MCMC efficiency and uncover the true simulated causal variables, including a gene-gene interaction. We applied PEAK to a case-control study of childhood asthma with 2,521 genetic variants. We used an informative graph for oxidative stress derived from Gene Ontology and identified several variants in ERBB4, OXR1, and BCL2 with strong evidence for associations with childhood asthma.
We introduced an extremely flexible analysis framework capable of efficiently performing Bayesian variable selection on many candidate variables. The PEAK algorithm can be provided with an informative graph, which can be advantageous when considering gene-gene interactions, or a symmetric graph, which simply divides the model space into manageable regions. The PEAK framework is compatible with various model forms, allowing for the algorithm to be configured for different study designs and applications, such as pathway or rare-variant analyses, by simple modifications to the model likelihood and proposal functions.
对众多遗传变异与疾病之间的边缘关联进行检测可能会忽略变量之间的复杂关系(例如,基因-基因相互作用)。贝叶斯方法可以一起对多个变量进行建模,并提供优于传统建模策略的优势,包括将现有生物学证据用作建模先验知识,并承认许多模型可能很好地适用于数据。对于许多候选变量,贝叶斯变量选择方法依赖于算法来近似模型的后验分布,例如马尔可夫链蒙特卡罗(MCMC)。不幸的是,MCMC 难以并行化,并且需要多次迭代才能充分采样后验。我们引入了一种可扩展的算法,称为 PEAK,该算法通过使用类似于山顶的有根图将一大组变量划分为相关组,从而提高了 MCMC 的效率。我们的算法在可用时利用了并行计算和现有的生物学数据库。
通过使用图来管理具有超过 500,000 个候选变量的模型空间,我们能够提高 MCMC 的效率,并发现真正的模拟因果变量,包括基因-基因相互作用。我们将 PEAK 应用于一项涉及 2521 个遗传变异的儿童哮喘病例对照研究。我们使用了来自基因本体论的氧化应激信息图,并鉴定出 ERBB4、OXR1 和 BCL2 中的几个变体与儿童哮喘有很强的关联证据。
我们引入了一种极其灵活的分析框架,能够在许多候选变量上高效执行贝叶斯变量选择。PEAK 算法可以提供一个信息图,当考虑基因-基因相互作用时,这可能是有利的,或者提供一个对称图,它只是将模型空间划分为可管理的区域。PEAK 框架与各种模型形式兼容,允许通过对模型似然和提议函数进行简单修改,将算法配置为不同的研究设计和应用,例如途径或罕见变异分析。