Bartha István, Carlson Jonathan M, Brumme Chanson J, McLaren Paul J, Brumme Zabrina L, John Mina, Haas David W, Martinez-Picado Javier, Dalmau Judith, López-Galíndez Cecilio, Casado Concepción, Rauch Andri, Günthard Huldrych F, Bernasconi Enos, Vernazza Pietro, Klimkait Thomas, Yerly Sabine, O'Brien Stephen J, Listgarten Jennifer, Pfeifer Nico, Lippert Christoph, Fusi Nicolo, Kutalik Zoltán, Allen Todd M, Müller Viktor, Harrigan P Richard, Heckerman David, Telenti Amalio, Fellay Jacques
School of Life Sciences , École Polytechnique Fédérale de Lausanne , Lausanne , Switzerland ; Institute of Microbiology , University Hospital and University of Lausanne , Lausanne , Switzerland ; Research Group of Theoretical Biology and Evolutionary Ecology , Eötvös Loránd University and the Hungarian Academy of Sciences , Budapest , Hungary ; Swiss Institute of Bioinformatics , Lausanne , Switzerland.
Elife. 2013 Oct 29;2:e01123. doi: 10.7554/eLife.01123.
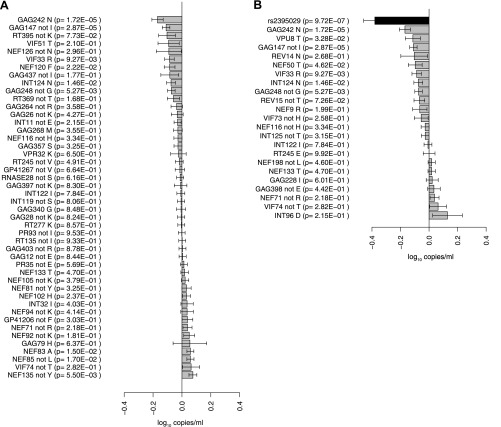
HIV-1 sequence diversity is affected by selection pressures arising from host genomic factors. Using paired human and viral data from 1071 individuals, we ran >3000 genome-wide scans, testing for associations between host DNA polymorphisms, HIV-1 sequence variation and plasma viral load (VL), while considering human and viral population structure. We observed significant human SNP associations to a total of 48 HIV-1 amino acid variants (p<2.4 × 10(-12)). All associated SNPs mapped to the HLA class I region. Clinical relevance of host and pathogen variation was assessed using VL results. We identified two critical advantages to the use of viral variation for identifying host factors: (1) association signals are much stronger for HIV-1 sequence variants than VL, reflecting the 'intermediate phenotype' nature of viral variation; (2) association testing can be run without any clinical data. The proposed genome-to-genome approach highlights sites of genomic conflict and is a strategy generally applicable to studies of host-pathogen interaction. DOI:http://dx.doi.org/10.7554/eLife.01123.001.
HIV-1序列多样性受宿主基因组因素产生的选择压力影响。利用来自1071名个体的配对人类和病毒数据,我们进行了3000多次全基因组扫描,测试宿主DNA多态性、HIV-1序列变异与血浆病毒载量(VL)之间的关联,同时考虑人类和病毒群体结构。我们观察到总共48个HIV-1氨基酸变体与人类SNP存在显著关联(p<2.4×10^(-12))。所有相关的SNP都定位于HLA I类区域。利用VL结果评估宿主和病原体变异的临床相关性。我们确定了利用病毒变异来识别宿主因素的两个关键优势:(1)HIV-1序列变体的关联信号比VL强得多,这反映了病毒变异的“中间表型”性质;(2)关联测试可以在没有任何临床数据的情况下进行。所提出的基因组对基因组方法突出了基因组冲突位点,是一种普遍适用于宿主-病原体相互作用研究的策略。DOI:http://dx.doi.org/10.7554/eLife.01123.001