New England Biolabs Inc., Ipswich, Massachusetts, United States of America.
PLoS One. 2013 Oct 28;8(10):e76096. doi: 10.1371/journal.pone.0076096. eCollection 2013.
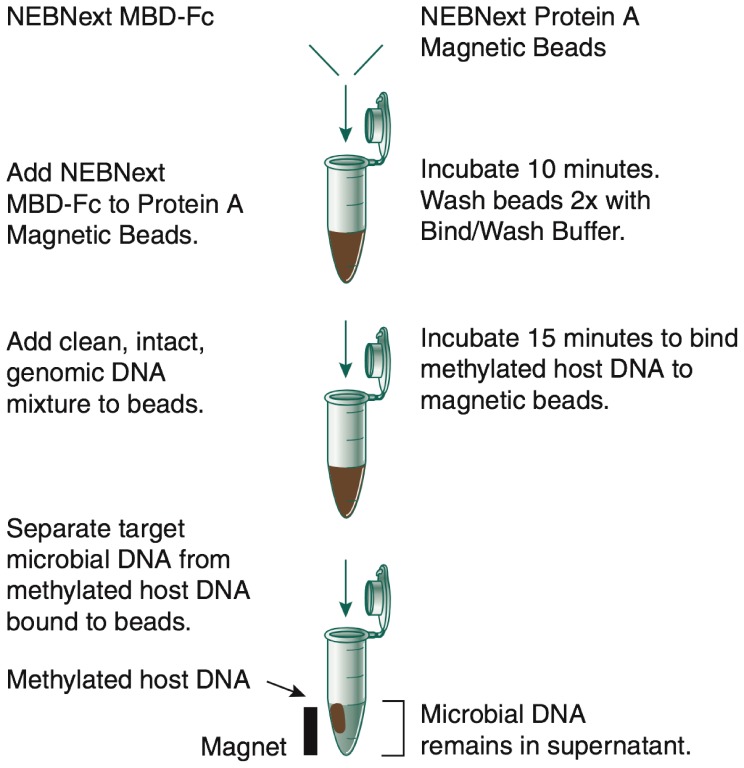
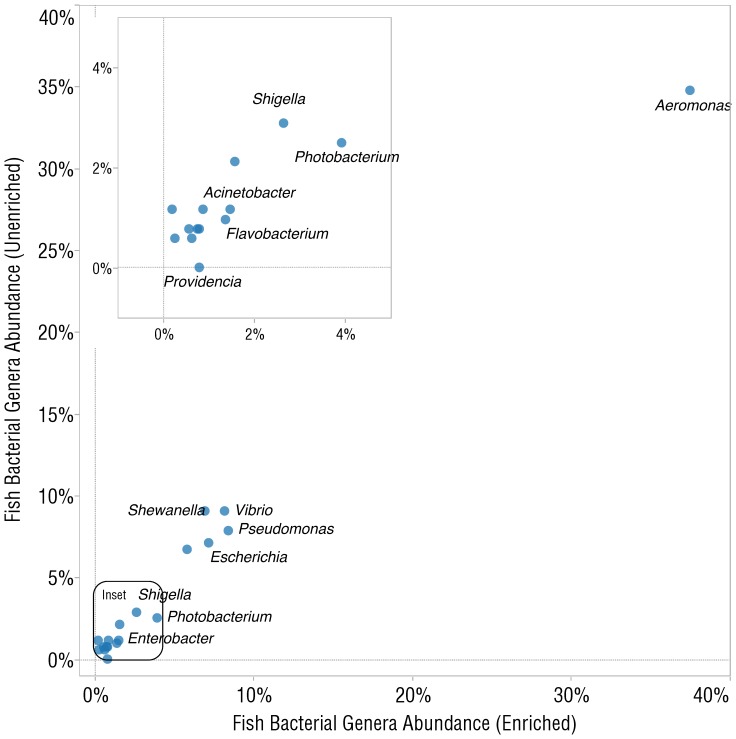
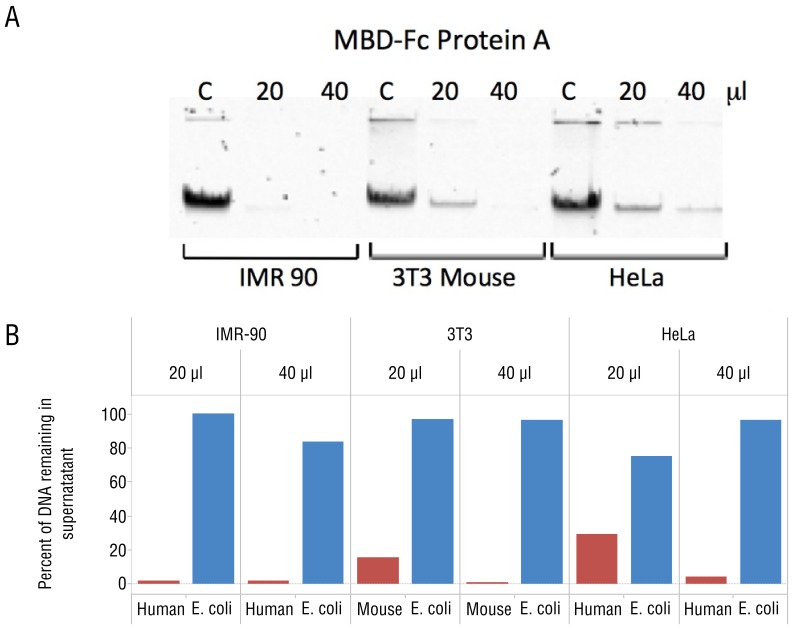
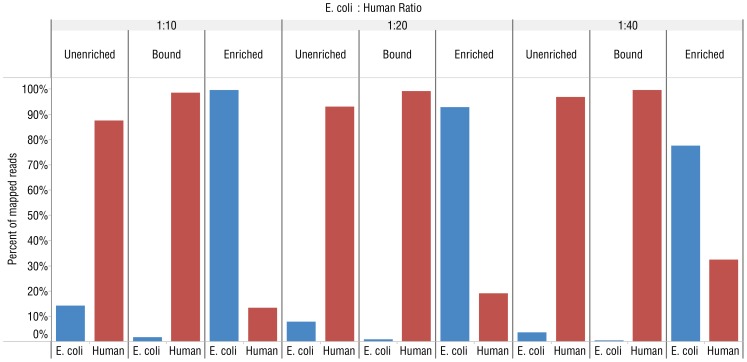
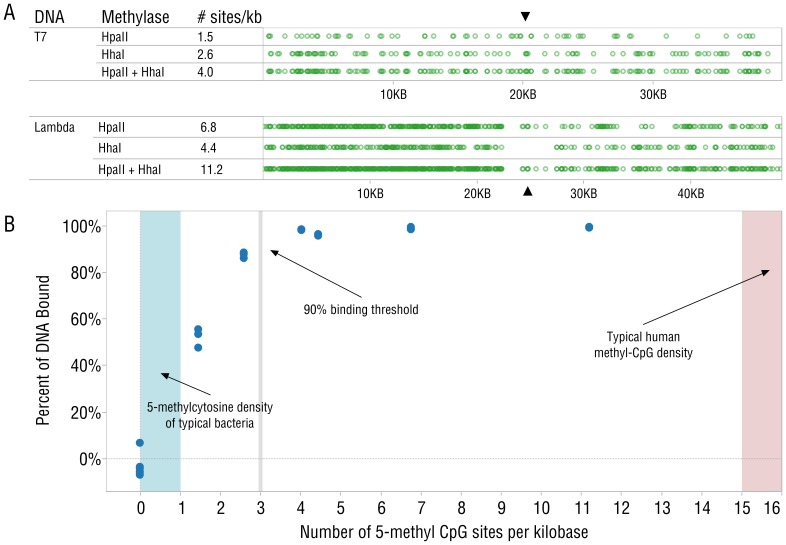
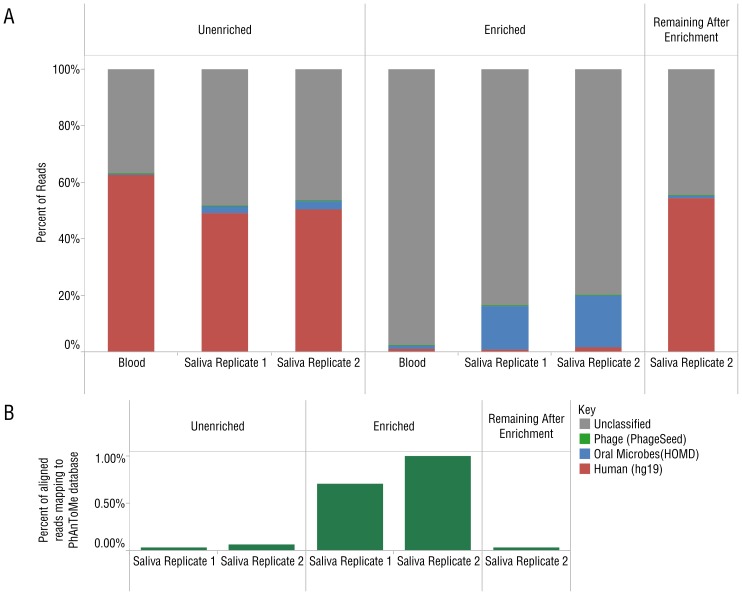
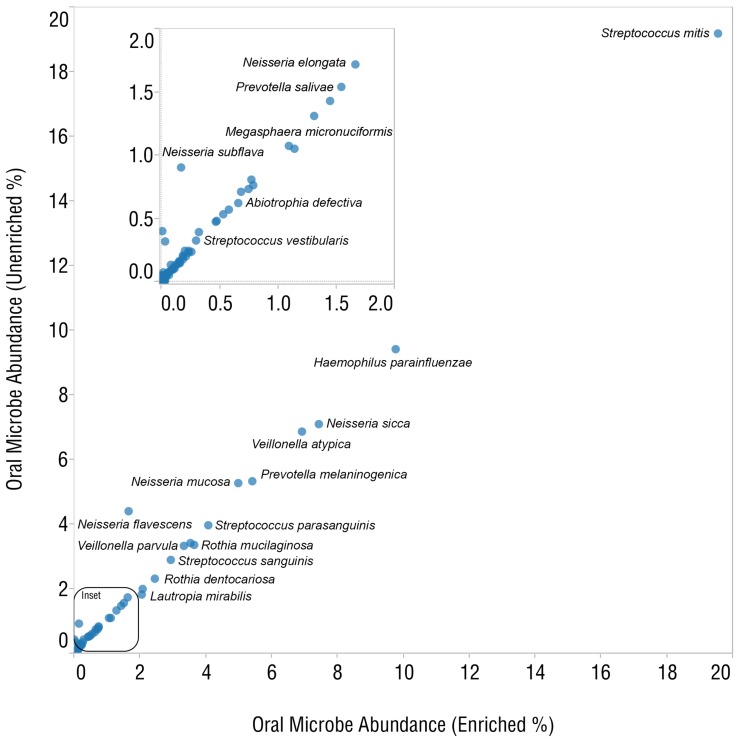
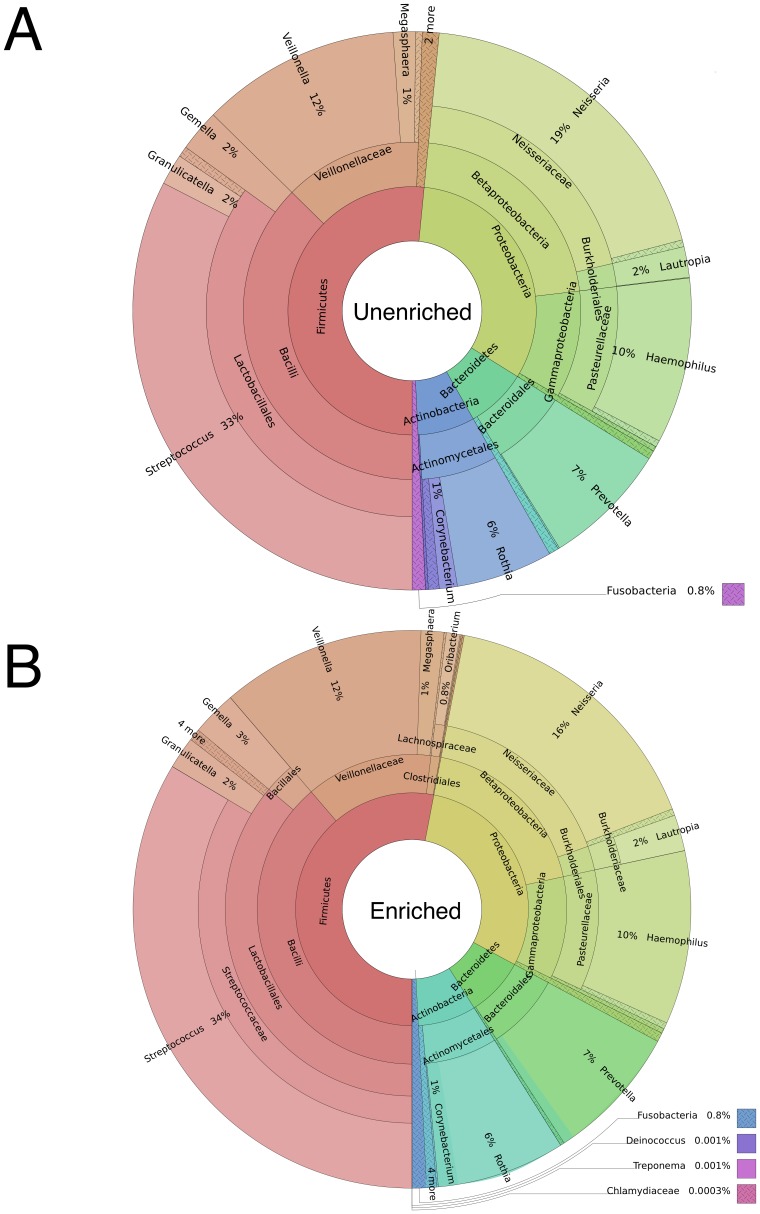
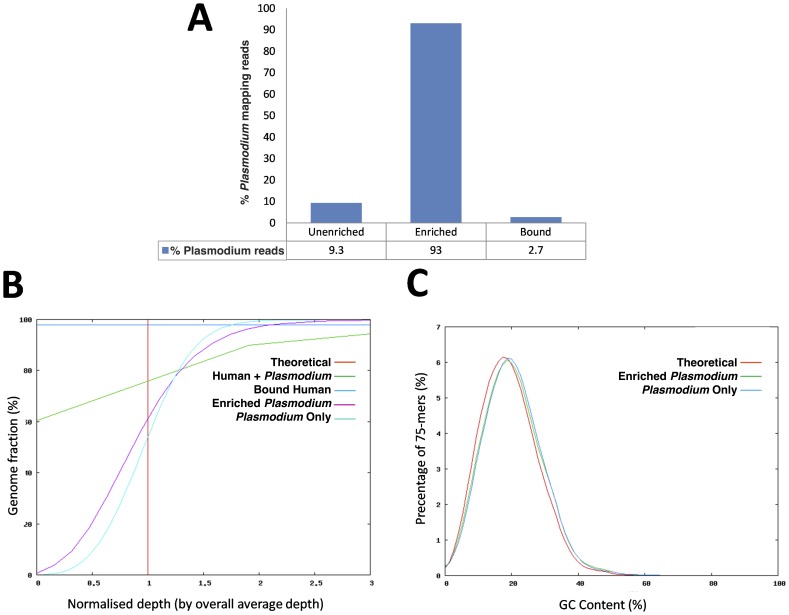
DNA samples derived from vertebrate skin, bodily cavities and body fluids contain both host and microbial DNA; the latter often present as a minor component. Consequently, DNA sequencing of a microbiome sample frequently yields reads originating from the microbe(s) of interest, but with a vast excess of host genome-derived reads. In this study, we used a methyl-CpG binding domain (MBD) to separate methylated host DNA from microbial DNA based on differences in CpG methylation density. MBD fused to the Fc region of a human antibody (MBD-Fc) binds strongly to protein A paramagnetic beads, forming an effective one-step enrichment complex that was used to remove human or fish host DNA from bacterial and protistan DNA for subsequent sequencing and analysis. We report enrichment of DNA samples from human saliva, human blood, a mock malaria-infected blood sample and a black molly fish. When reads were mapped to reference genomes, sequence reads aligning to host genomes decreased 50-fold, while bacterial and Plasmodium DNA sequences reads increased 8-11.5-fold. The Shannon-Wiener diversity index was calculated for 149 bacterial species in saliva before and after enrichment. Unenriched saliva had an index of 4.72, while the enriched sample had an index of 4.80. The similarity of these indices demonstrates that bacterial species diversity and relative phylotype abundance remain conserved in enriched samples. Enrichment using the MBD-Fc method holds promise for targeted microbiome sequence analysis across a broad range of sample types.
从脊椎动物皮肤、体腔和体液中提取的 DNA 样本既含有宿主 DNA 又含有微生物 DNA;后者通常作为次要成分存在。因此,对微生物组样本进行 DNA 测序通常会产生源自感兴趣微生物的读段,但宿主基因组衍生的读段数量要多得多。在这项研究中,我们使用甲基化 CpG 结合域(MBD)根据 CpG 甲基化密度的差异,将甲基化的宿主 DNA 与微生物 DNA 分离。MBD 与人类抗体的 Fc 区融合(MBD-Fc)与蛋白 A 顺磁珠结合牢固,形成有效的一步富集复合物,用于从细菌和原生动物 DNA 中去除人类或鱼类宿主 DNA,以便随后进行测序和分析。我们报告了从人唾液、人血、模拟疟疾感染血样和黑玛丽鱼中分离的 DNA 样本的富集情况。当将读段映射到参考基因组时,与宿主基因组对齐的序列读段减少了 50 倍,而细菌和疟原虫 DNA 序列读段增加了 8-11.5 倍。对未富集和富集后的唾液中的 149 种细菌进行了 Shannon-Wiener 多样性指数计算。未富集的唾液指数为 4.72,而富集后的样本指数为 4.80。这些指数的相似性表明,在富集样本中,细菌物种多样性和相对系统发育丰度保持不变。MBD-Fc 方法的富集有望在广泛的样本类型中进行靶向微生物组序列分析。