School of Biological Sciences, Centre for Brain Research, The University of Auckland, Auckland, New Zealand.
Sci Rep. 2020 Nov 6;10(1):19255. doi: 10.1038/s41598-020-76022-4.
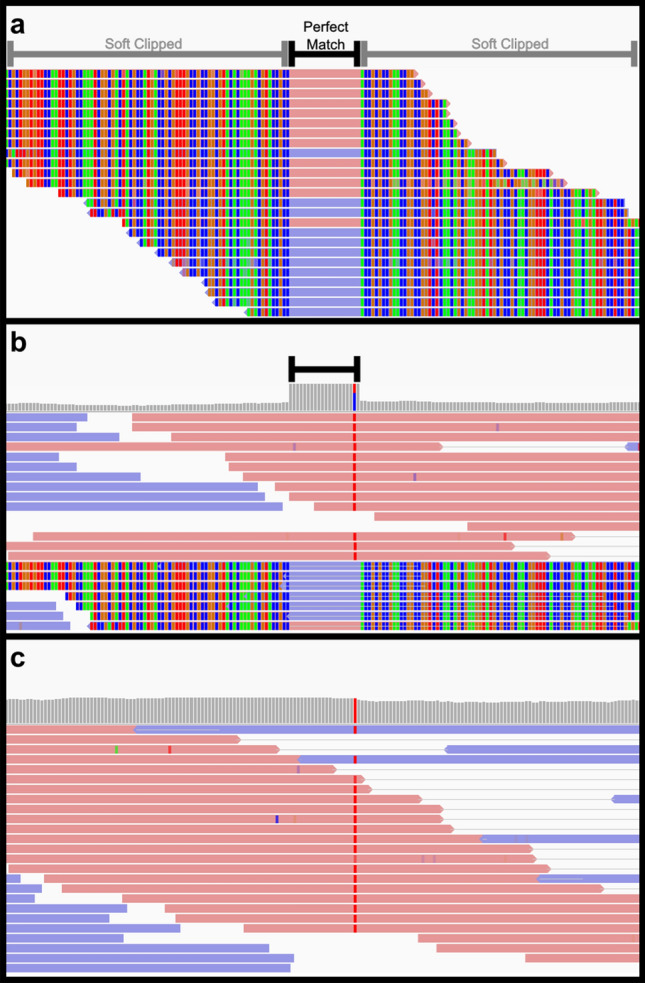
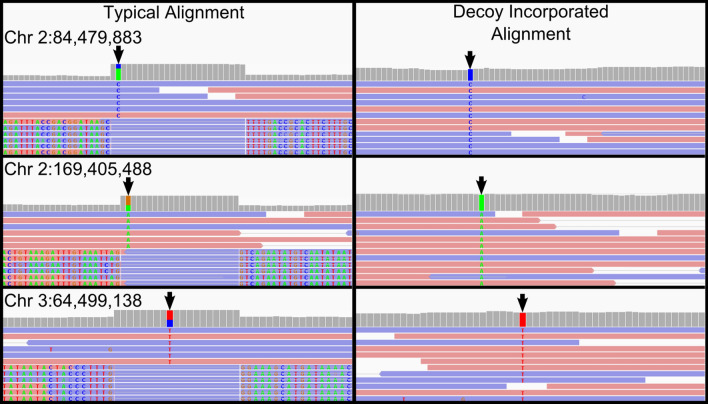
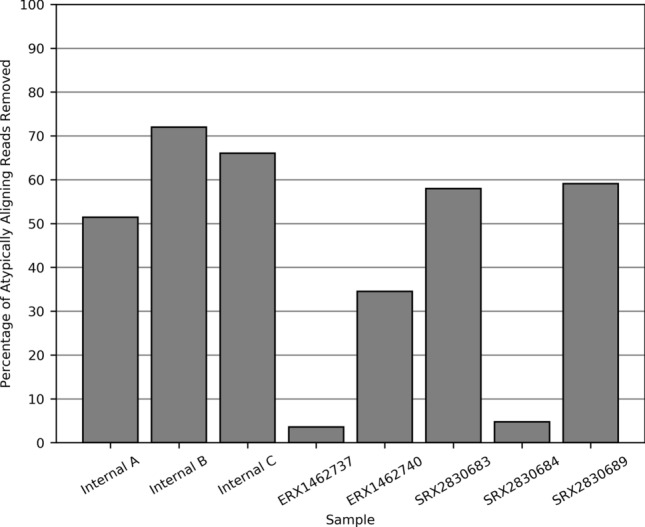
Cells obtained from human saliva are commonly used as an alternative DNA source when blood is difficult or less convenient to collect. Although DNA extracted from saliva is considered to be of comparable quality to that derived from blood, recent studies have shown that non-human contaminating DNA derived from saliva can confound whole genome sequencing results. The most concerning complication is that non-human reads align to the human reference genome using standard methodology, which can critically affect the resulting variant genotypes identified in a genome. We identified clusters of anomalous variants in saliva DNA derived reads which aligned in an atypical manner. These reads had only short regions of identity to the human reference sequence, flanked by soft clipped sequence. Sequence comparisons of atypically aligning reads from eight human saliva-derived samples to RefSeq genomes revealed the majority to be of bacterial origin (63.46%). To partition the non-human reads during the alignment step, a decoy of the most prevalent bacterial genome sequences was designed and utilised. This reduced the number of atypically aligning reads when trialled on the eight saliva-derived samples by 44% and most importantly prevented the associated anomalous genotype calls. Saliva derived DNA is often contaminated by DNA from other species. This can lead to non-human reads aligning to the human reference genome using current alignment best-practices, impacting variant identification. This problem can be diminished by using a bacterial decoy in the alignment process.
从人类唾液中获得的细胞通常被用作血液难以或不方便采集时的替代 DNA 来源。虽然从唾液中提取的 DNA 被认为与血液中提取的 DNA 质量相当,但最近的研究表明,来自唾液的非人类污染 DNA 会混淆全基因组测序结果。最令人担忧的并发症是,使用标准方法,非人类的读取可以与人类参考基因组对齐,这会严重影响基因组中识别出的变体基因型。我们在以非典型方式对齐的唾液 DNA 衍生读取中发现了异常变异簇。这些读取与人类参考序列只有很短的同源区域,两侧是软剪辑序列。对来自八个人类唾液样本的异常对齐读取与 RefSeq 基因组进行序列比较,发现大多数来自细菌(63.46%)。为了在对齐步骤中将非人类读取分开,设计并使用了最常见的细菌基因组序列的诱饵。当在八个唾液衍生样本上进行试验时,这减少了异常对齐读取的数量 44%,最重要的是,防止了相关的异常基因型调用。唾液 DNA 经常受到其他物种 DNA 的污染。这可能导致非人类的读取使用当前的对齐最佳实践与人类参考基因组对齐,从而影响变异的识别。通过在对齐过程中使用细菌诱饵,可以减少这个问题。