Cojocaru Corneliu, Airinei Anton, Fifere Nicusor
"Petru Poni" Institute of Macromolecular Chemistry, Aleea Grigore Ghica Voda 41A, 700487 Iasi, Romania.
Springerplus. 2013 Oct 31;2(1):586. doi: 10.1186/2193-1801-2-586. eCollection 2013.
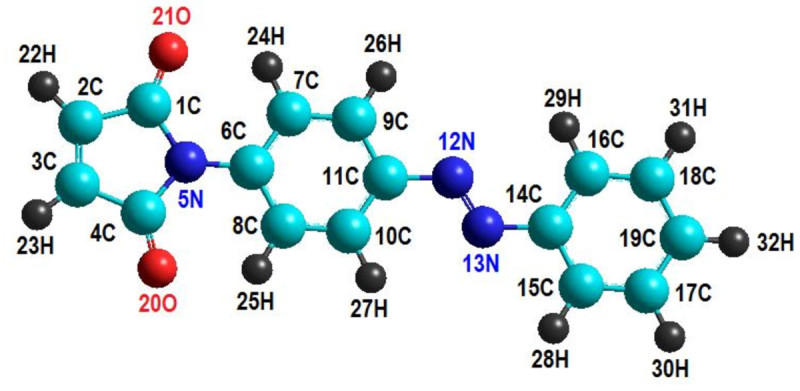
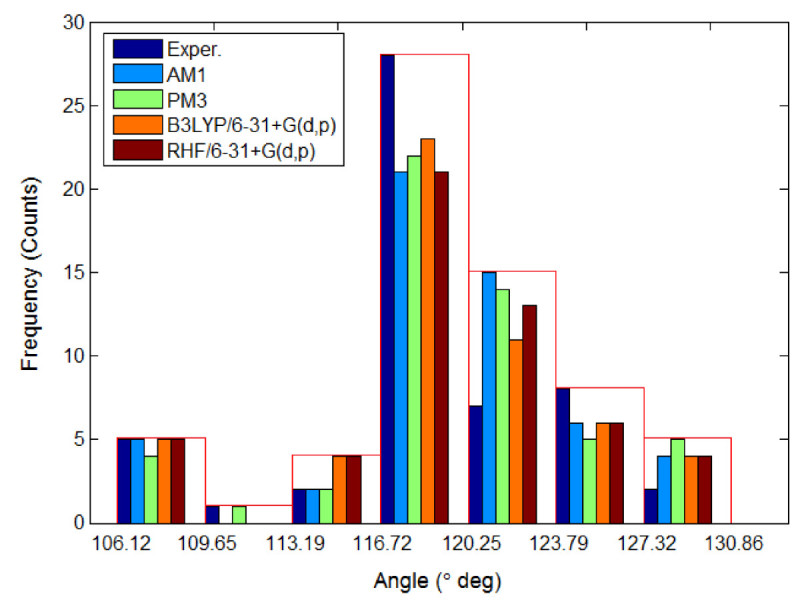
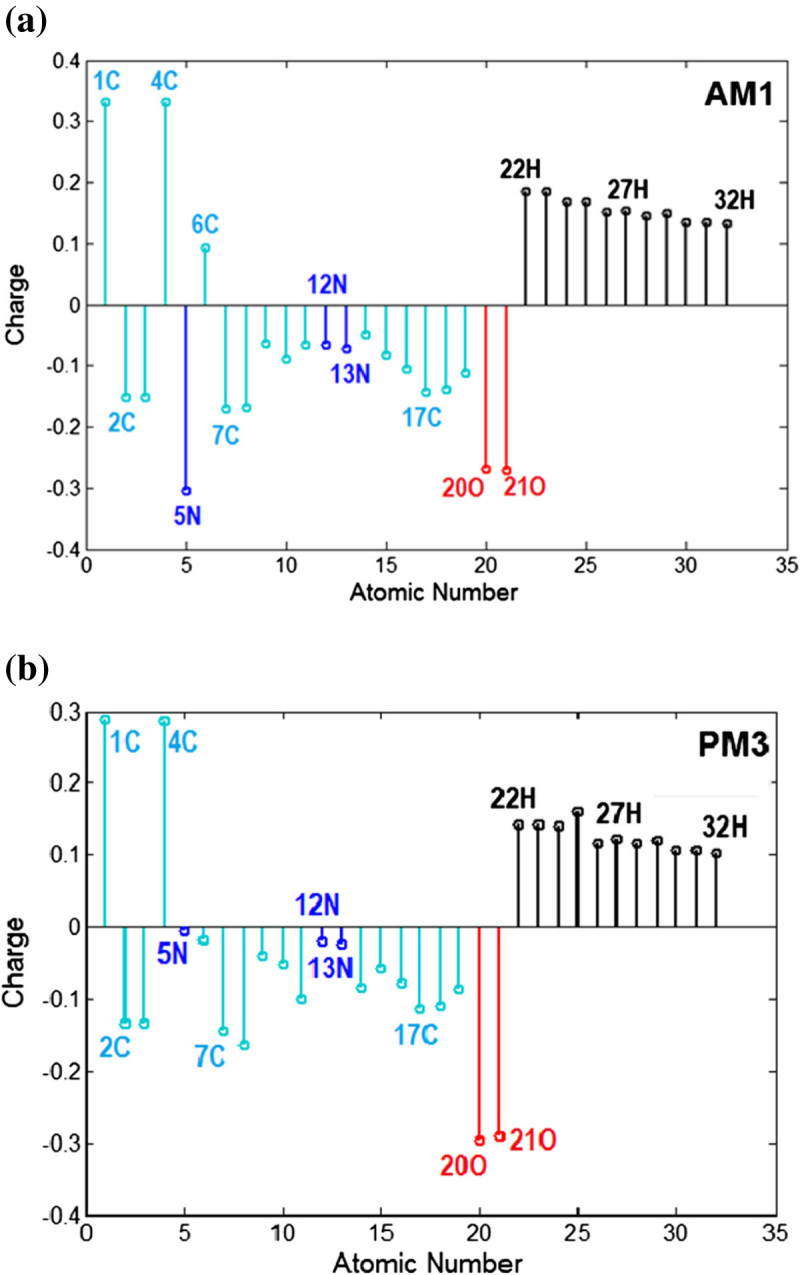
The molecular orbital calculations have been carried out to investigate the structure and stability of (E) / (Z) isomers of some azobenzene derivatives containing maleimide groups. A special attention has been devoted to the compound (E)-1, (E)-1-(4-(phenyldiazenyl)phenyl)-1H-pyrrole-2,5-dione, for which the available crystallographic experimental data have been used to validate the modeling structures computed at the theoretical levels AM1, PM3, RHF/6-31+G(d,p) and B3LYP/6-31+G(d,p). To this end, the discrepancy between experimental and calculated structural parameters has been ascertained in terms of root-mean-square deviation (RMSD). The quantum calculations at the level RHF/6-31+G(d,p) yield the most accurate results on (E)-1 structure giving a deviation error from crystallographic data of about 5.00% for bond lengths and 0.97% for interatomic angles. The theoretical electronic absorption spectra of azobenzene derivatives of concern have been computed by means of configuration-interaction method (CI) at the level of semi-empirical Hamiltonians (AM1 and PM3). Likewise, the molecular energy spectra, electrostatic potential and some quantitative structure activity relationship (QSAR) properties of studied molecules have been computed and discussed in the paper.
已进行分子轨道计算,以研究一些含马来酰亚胺基团的偶氮苯衍生物的(E)/(Z)异构体的结构和稳定性。特别关注了化合物(E)-1,即(E)-1-(4-(苯基重氮基)苯基)-1H-吡咯-2,5-二酮,已使用现有的晶体学实验数据来验证在理论水平AM1、PM3、RHF/6-31+G(d,p)和B3LYP/6-31+G(d,p)下计算的模型结构。为此,已根据均方根偏差(RMSD)确定了实验和计算结构参数之间的差异。在RHF/6-31+G(d,p)水平上的量子计算得出了关于(E)-1结构的最准确结果,其键长与晶体学数据的偏差误差约为5.00%,原子间角度的偏差误差为0.97%。通过在半经验哈密顿量(AM1和PM3)水平上的组态相互作用方法(CI)计算了相关偶氮苯衍生物的理论电子吸收光谱。同样,本文还计算并讨论了所研究分子的分子能谱、静电势和一些定量构效关系(QSAR)性质。