Hollie Norris I, Cash James G, Matlib M Abdul, Wortman Matthew, Basford Joshua E, Abplanalp William, Hui David Y
Department of Pathology and Laboratory Medicine, Metabolic Diseases Institute, University of Cincinnati College of Medicine, Cincinnati, OH, USA.
Department of Pharmacology and Cell Biophysics, University of Cincinnati College of Medicine, Cincinnati, OH, USA.
Biochim Biophys Acta. 2014 Jun;1841(6):888-95. doi: 10.1016/j.bbalip.2013.11.013. Epub 2013 Dec 5.
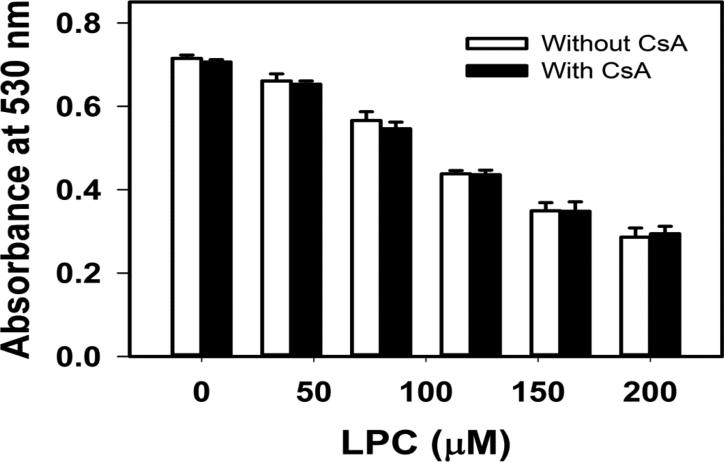
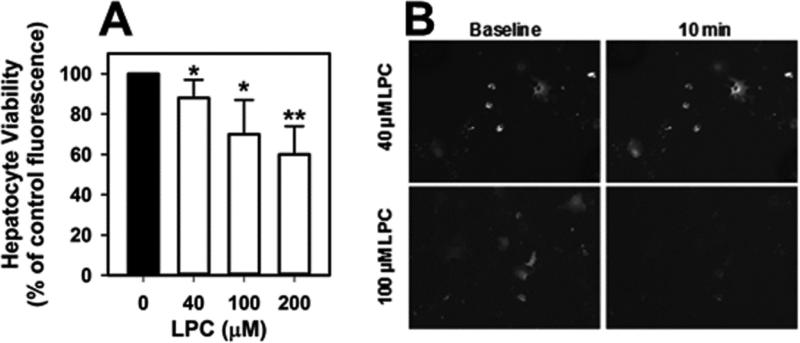
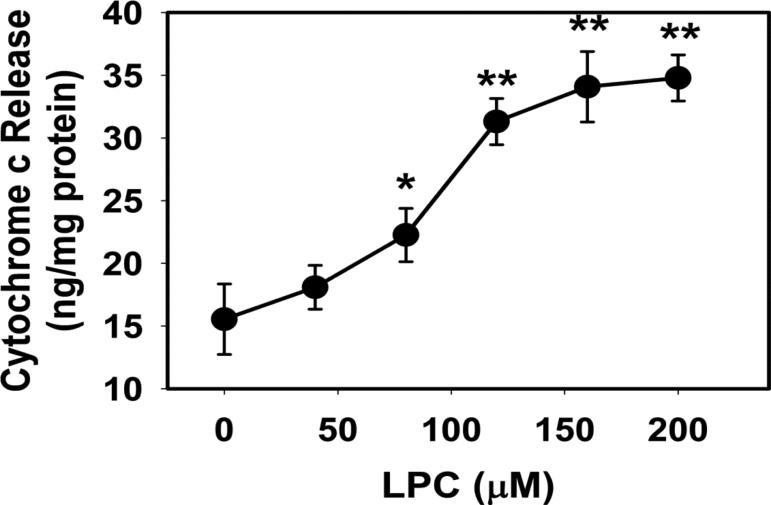
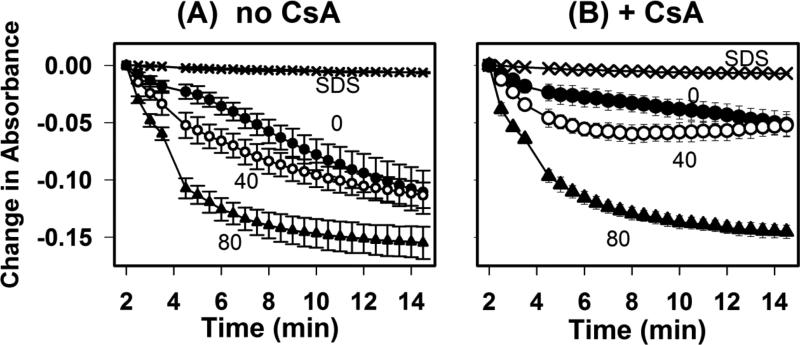
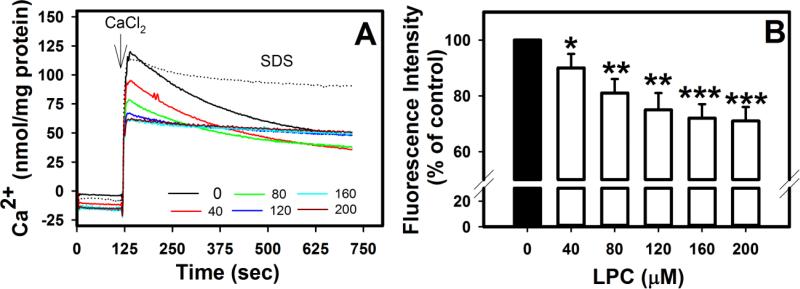
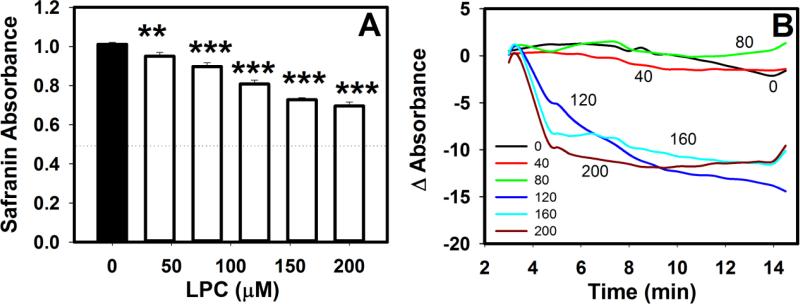
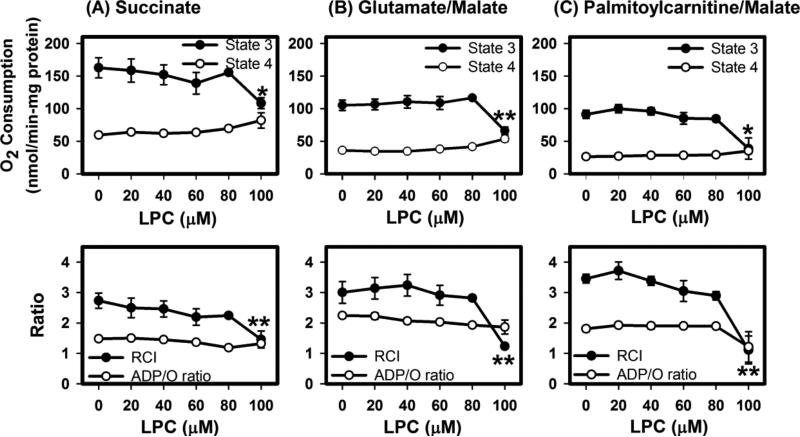
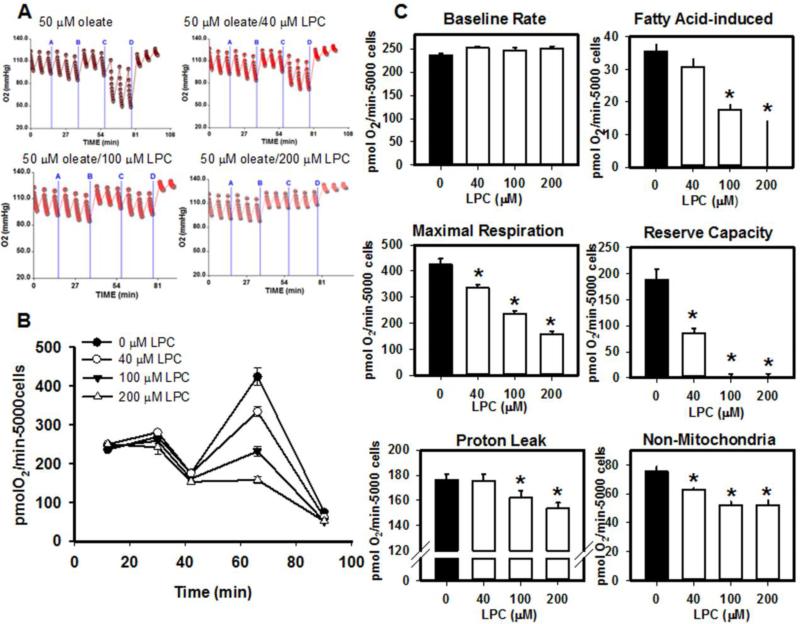
Mice deficient in group 1b phospholipase A2 have decreased plasma lysophosphatidylcholine and increased hepatic oxidation that is inhibited by intraperitoneal lysophosphatidylcholine injection. This study sought to identify a mechanism for lysophosphatidylcholine-mediated inhibition of hepatic oxidative function. Results showed that in vitro incubation of isolated mitochondria with 40-200μM lysophosphatidylcholine caused cyclosporine A-resistant swelling in a concentration-dependent manner. However, when mitochondria were challenged with 220μM CaCl2, cyclosporine A protected against permeability transition induced by 40μM, but not 80μM lysophosphatidylcholine. Incubation with 40-120μM lysophosphatidylcholine also increased mitochondrial permeability to 75μM CaCl2 in a concentration-dependent manner. Interestingly, despite incubation with 80μM lysophosphatidylcholine, the mitochondrial membrane potential was steady in the presence of succinate, and oxidation rates and respiratory control indices were similar to controls in the presence of succinate, glutamate/malate, and palmitoyl-carnitine. However, mitochondrial oxidation rates were inhibited by 30-50% at 100μM lysophosphatidylcholine. Finally, while 40μM lysophosphatidylcholine has no effect on fatty acid oxidation and mitochondria remained impermeable in intact hepatocytes, 100μM lysophosphatidylcholine inhibited fatty acid stimulated oxidation and caused intracellular mitochondrial permeability. Taken together, these present data demonstrated that LPC concentration dependently modulates mitochondrial microenvironment, with low micromolar concentrations of lysophosphatidylcholine sufficient to change hepatic oxidation rate whereas higher concentrations are required to disrupt mitochondrial integrity.
缺乏1b组磷脂酶A2的小鼠血浆溶血磷脂酰胆碱水平降低,肝脏氧化增加,腹腔注射溶血磷脂酰胆碱可抑制这种增加。本研究旨在确定溶血磷脂酰胆碱介导的肝脏氧化功能抑制机制。结果表明,分离的线粒体与40 - 200μM溶血磷脂酰胆碱在体外孵育会导致环孢素A耐药性肿胀,且呈浓度依赖性。然而,当线粒体用220μM氯化钙刺激时,环孢素A可保护其免受40μM而非80μM溶血磷脂酰胆碱诱导的通透性转变。与40 - 120μM溶血磷脂酰胆碱孵育也会使线粒体对75μM氯化钙的通透性呈浓度依赖性增加。有趣的是,尽管与80μM溶血磷脂酰胆碱孵育,但在琥珀酸存在下线粒体膜电位稳定,在琥珀酸、谷氨酸/苹果酸和棕榈酰肉碱存在下氧化速率和呼吸控制指数与对照相似。然而,在100μM溶血磷脂酰胆碱时线粒体氧化速率受到30 - 50%的抑制。最后,虽然40μM溶血磷脂酰胆碱对脂肪酸氧化无影响,且完整肝细胞中的线粒体仍保持不透性,但100μM溶血磷脂酰胆碱会抑制脂肪酸刺激的氧化并导致细胞内线粒体通透性增加。综上所述,这些现有数据表明溶血磷脂酰胆碱浓度依赖性地调节线粒体微环境,低微摩尔浓度的溶血磷脂酰胆碱足以改变肝脏氧化速率,而需要更高浓度才能破坏线粒体完整性。