Xia Kelin, Feng Xin, Chen Zhan, Tong Yiying, Wei Guo Wei
Department of Mathematics, Michigan State University, MI 48824, USA.
J Comput Phys. 2014 Jan;257(Pt A). doi: 10.1016/j.jcp.2013.09.034.
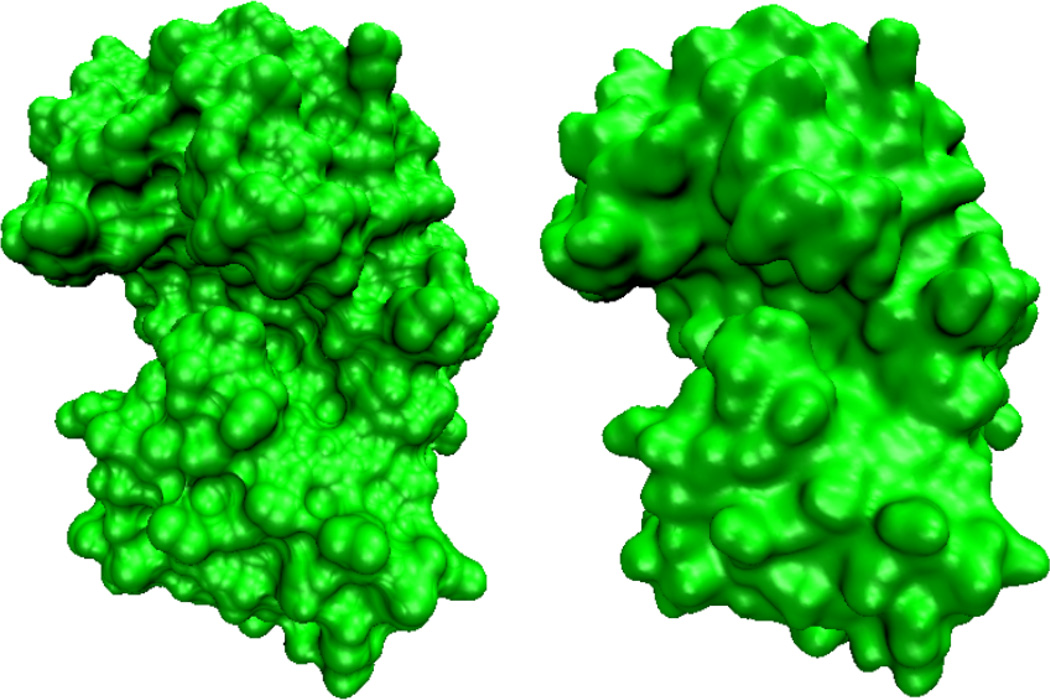
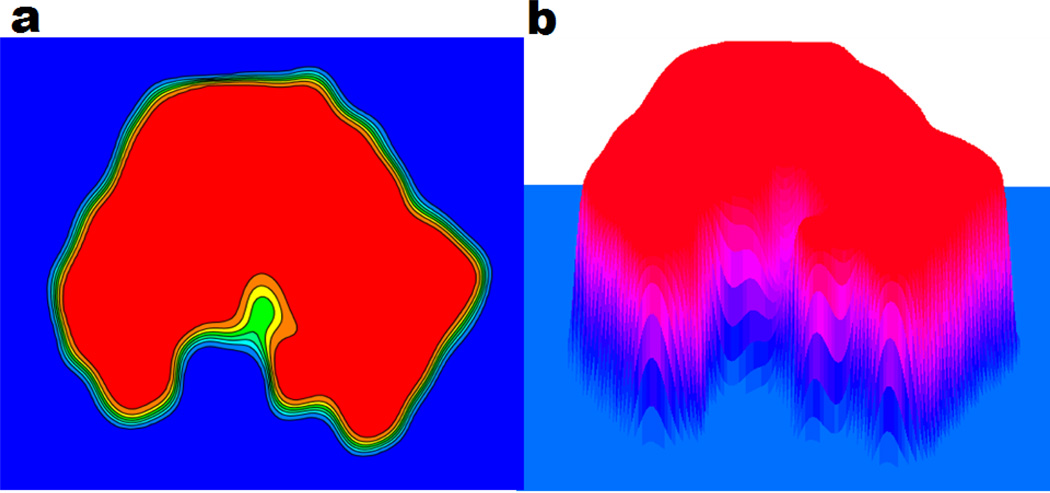
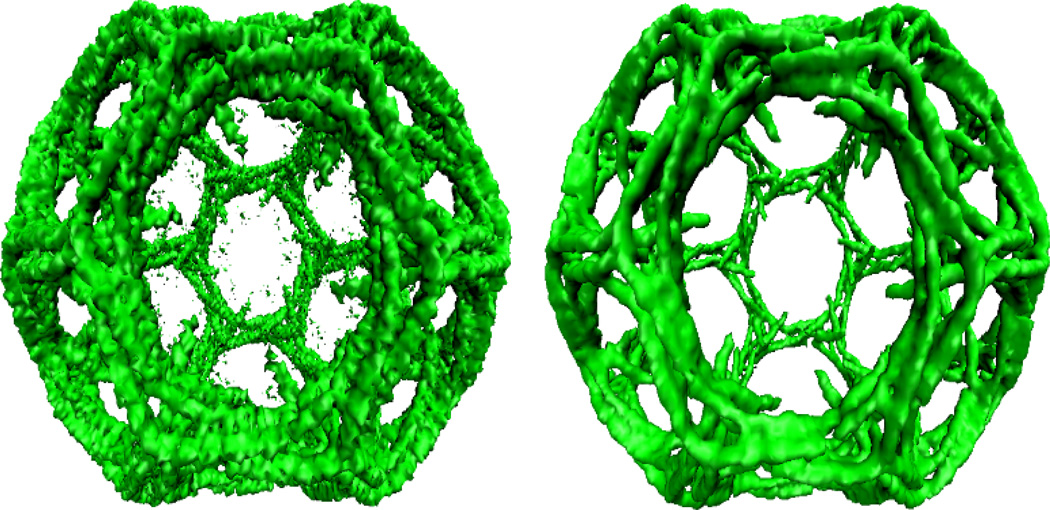
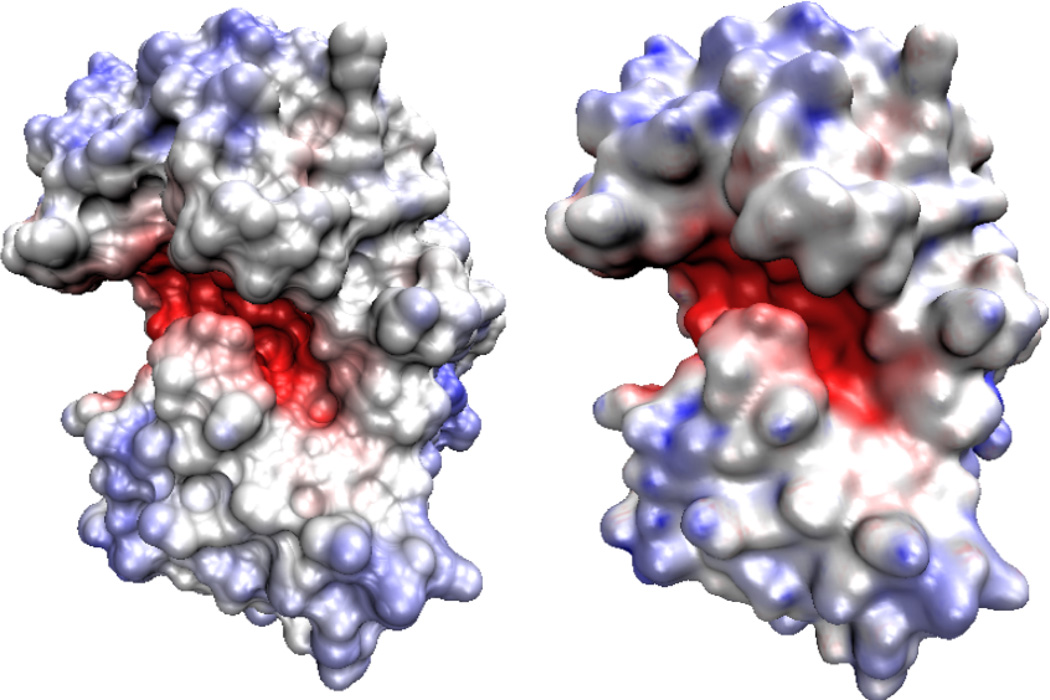
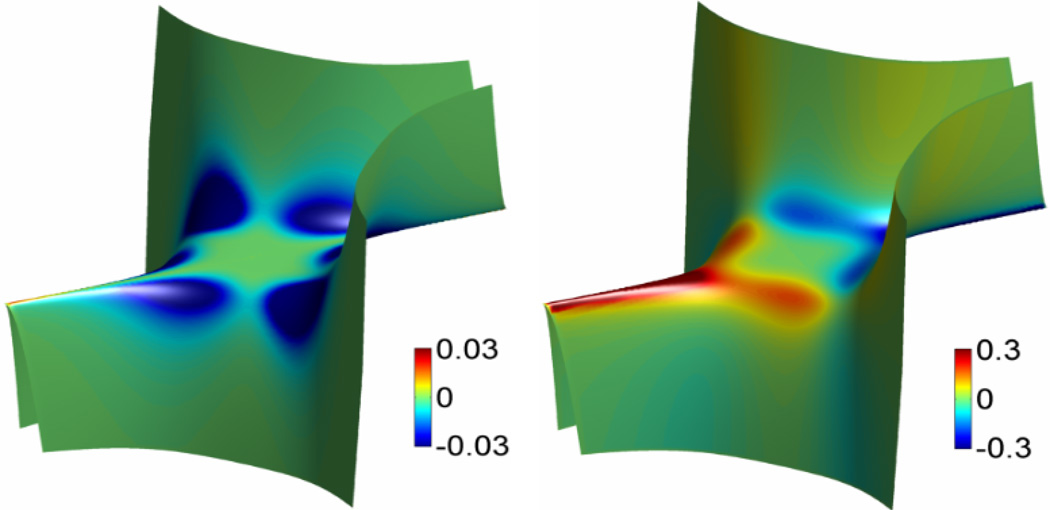
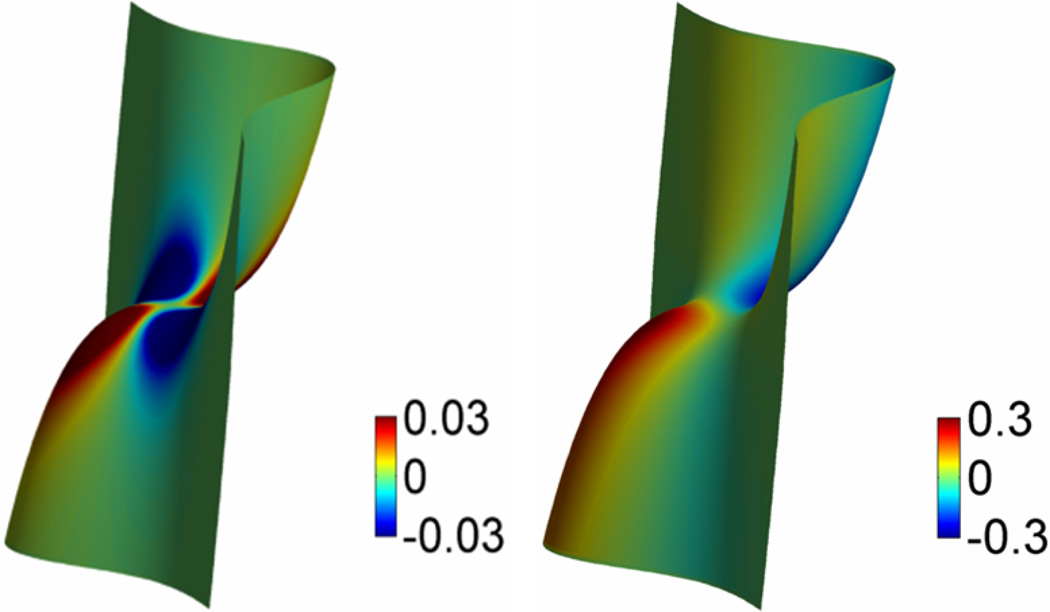
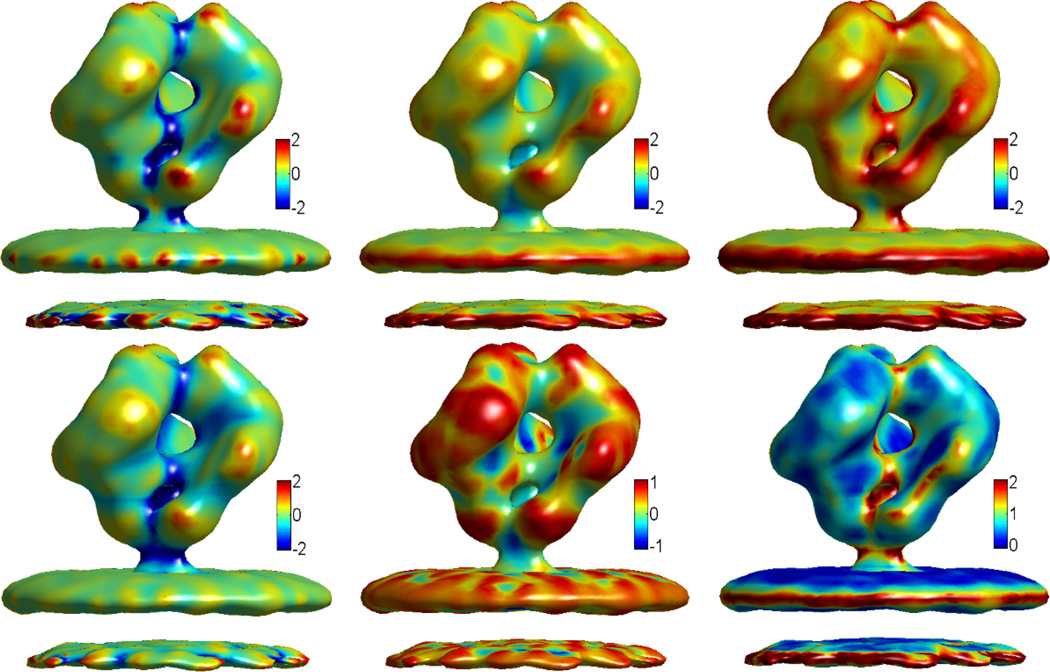
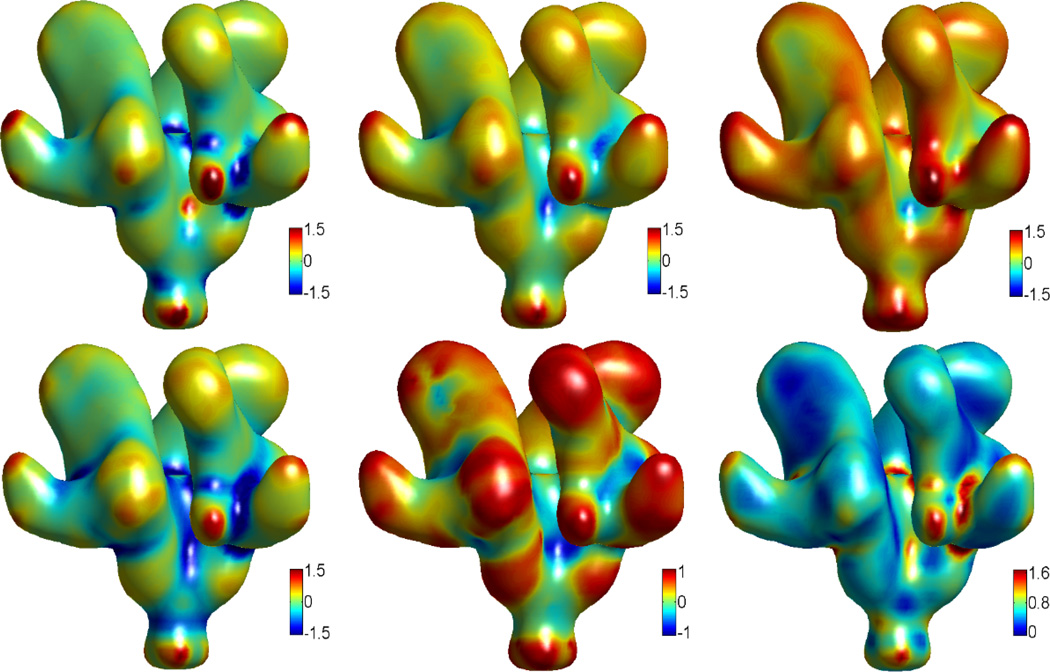
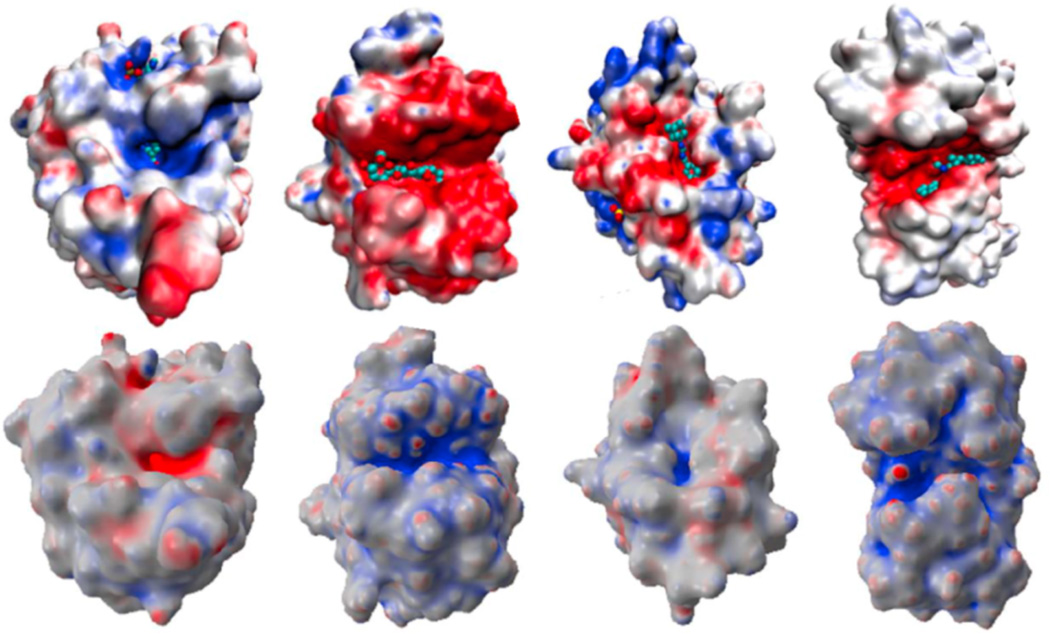
This paper focuses on the geometric modeling and computational algorithm development of biomolecular structures from two data sources: Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) in the Eulerian (or Cartesian) representation. Molecular surface (MS) contains non-smooth geometric singularities, such as cusps, tips and self-intersecting facets, which often lead to computational instabilities in molecular simulations, and violate the physical principle of surface free energy minimization. Variational multiscale surface definitions are proposed based on geometric flows and solvation analysis of biomolecular systems. Our approach leads to geometric and potential driven Laplace-Beltrami flows for biomolecular surface evolution and formation. The resulting surfaces are free of geometric singularities and minimize the total free energy of the biomolecular system. High order partial differential equation (PDE)-based nonlinear filters are employed for EMDB data processing. We show the efficacy of this approach in feature-preserving noise reduction. After the construction of protein multiresolution surfaces, we explore the analysis and characterization of surface morphology by using a variety of curvature definitions. Apart from the classical Gaussian curvature and mean curvature, maximum curvature, minimum curvature, shape index, and curvedness are also applied to macromolecular surface analysis for the first time. Our curvature analysis is uniquely coupled to the analysis of electrostatic surface potential, which is a by-product of our variational multiscale solvation models. As an expository investigation, we particularly emphasize the numerical algorithms and computational protocols for practical applications of the above multiscale geometric models. Such information may otherwise be scattered over the vast literature on this topic. Based on the curvature and electrostatic analysis from our multiresolution surfaces, we introduce a new concept, the polarized curvature, for the prediction of protein binding sites.
本文聚焦于从两个数据源(蛋白质数据库(PDB)和电子显微镜数据库(EMDB))以欧拉(或笛卡尔)表示法进行生物分子结构的几何建模和计算算法开发。分子表面(MS)包含非光滑的几何奇点,如尖点、尖端和自相交面,这常常导致分子模拟中的计算不稳定性,并违反表面自由能最小化的物理原理。基于生物分子系统的几何流和溶剂化分析,提出了变分多尺度表面定义。我们的方法导致了用于生物分子表面演化和形成的几何和势驱动的拉普拉斯 - 贝尔特拉米流。所得表面没有几何奇点,并使生物分子系统的总自由能最小化。基于高阶偏微分方程(PDE)的非线性滤波器用于EMDB数据处理。我们展示了这种方法在保留特征的降噪方面的有效性。在构建蛋白质多分辨率表面之后,我们通过使用各种曲率定义来探索表面形态的分析和表征。除了经典的高斯曲率和平均曲率外,最大曲率、最小曲率、形状指数和曲率也首次应用于大分子表面分析。我们的曲率分析与静电表面势的分析独特地耦合在一起,静电表面势是我们变分多尺度溶剂化模型的一个副产品。作为一项解释性研究,我们特别强调上述多尺度几何模型实际应用的数值算法和计算协议。否则,此类信息可能分散在关于该主题的大量文献中。基于我们多分辨率表面的曲率和静电分析,我们引入了一个新概念——极化曲率,用于预测蛋白质结合位点。