Livaja Maren, Wang Yu, Wieckhorst Silke, Haseneyer Grit, Seidel Michael, Hahn Volker, Knapp Steven J, Taudien Stefan, Schön Chris-Carolin, Bauer Eva
BMC Genomics. 2013 Sep 17;14:628. doi: 10.1186/1471-2164-14-628.
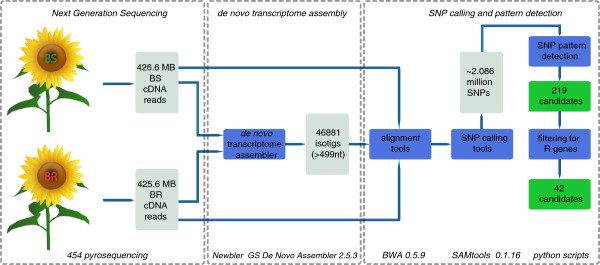
Sunflower belongs to the largest plant family on earth, the genomically poorly explored Compositae. Downy mildew Plasmopara halstedii (Farlow) Berlese & de Toni is one of the major diseases of cultivated sunflower (Helianthus annuus L.). In the search for new sources of downy mildew resistance, the locus Pl(ARG)on linkage group 1 (LG1) originating from H. argophyllus is promising since it confers resistance against all known races of the pathogen. However, the mapping resolution in the Pl(ARG) region is hampered by significantly suppressed recombination and by limited availability of polymorphic markers. Here we examined a strategy developed for the enrichment of molecular markers linked to this specific genomic region. We combined bulked segregant analysis (BSA) with next-generation sequencing (NGS) and de novo assembly of the sunflower transcriptome for single nucleotide polymorphism (SNP) discovery in a sequence resource combining reads originating from two sunflower species, H. annuus and H. argophyllus.
A computational pipeline developed for SNP calling and pattern detection identified 219 candidate genes. For a proof of concept, 42 resistance gene-like sequences were subjected to experimental SNP validation. Using a high-resolution mapping population, 12 SNP markers were mapped to LG1. We successfully verified candidate sequences either co-segregating with or closely flanking Pl(ARG).
This study is the first successful example to improve bulked segregant analysis with de novo transcriptome assembly using next generation sequencing. The BSTA pipeline we developed provides a useful guide for similar studies in other non-model organisms. Our results demonstrate this method is an efficient way to enrich molecular markers and to identify candidate genes in a specific mapping interval.
向日葵属于地球上最大的植物科——菊科,其基因组尚未得到充分研究。霜霉病(由寄生霜霉引起)是栽培向日葵(Helianthus annuus L.)的主要病害之一。在寻找霜霉病抗性新来源的过程中,源自银叶向日葵的1号连锁群(LG1)上的Pl(ARG)位点很有前景,因为它对该病原菌的所有已知小种都具有抗性。然而,Pl(ARG)区域的图谱分辨率受到显著抑制的重组以及多态性标记可用性有限的阻碍。在此,我们研究了一种用于富集与该特定基因组区域连锁的分子标记的策略。我们将混合分离群体分析法(BSA)与新一代测序(NGS)以及向日葵转录组的从头组装相结合,以便在一个结合了源自两个向日葵物种(向日葵和银叶向日葵)的reads的序列资源中发现单核苷酸多态性(SNP)。
一个为SNP检测和模式识别而开发的计算流程鉴定出219个候选基因。为了进行概念验证,对42个类抗性基因序列进行了实验性SNP验证。利用一个高分辨率的作图群体,12个SNP标记被定位到LG1上。我们成功验证了与Pl(ARG)共分离或紧密连锁的候选序列。
本研究是利用新一代测序进行从头转录组组装来改进混合分离群体分析法的首个成功实例。我们开发的BSTA流程为其他非模式生物的类似研究提供了有用的指导。我们的结果表明,这种方法是富集分子标记并在特定作图区间鉴定候选基因的有效途径。