Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, Massachusetts, United States of America.
Department of Biostatistics, Harvard School of Public Health, Boston, Massachusetts, United States of America.
PLoS Comput Biol. 2013;9(12):e1003392. doi: 10.1371/journal.pcbi.1003392. Epub 2013 Dec 12.
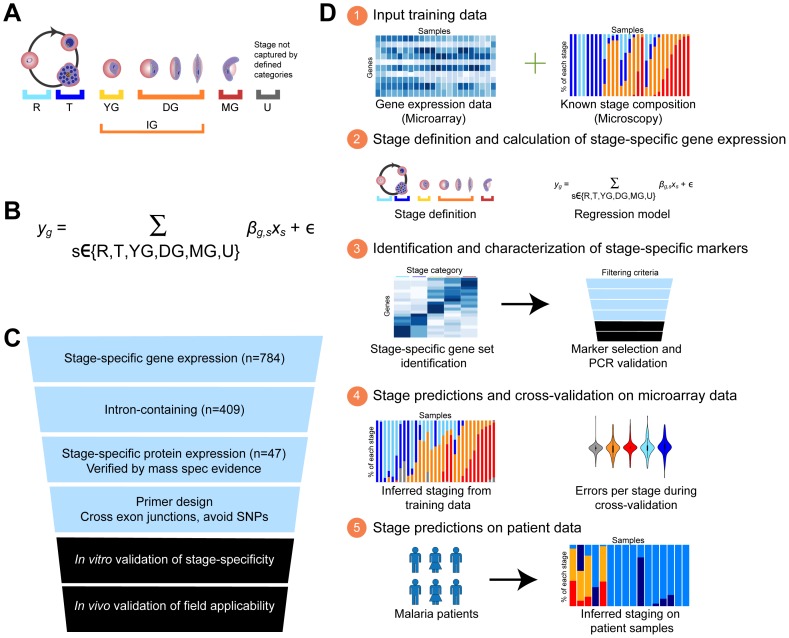
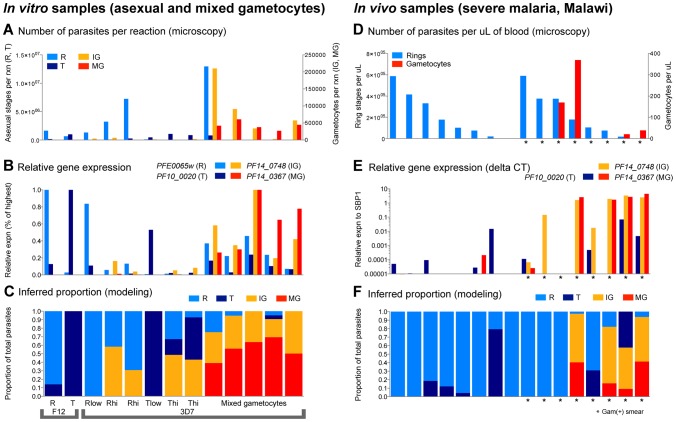
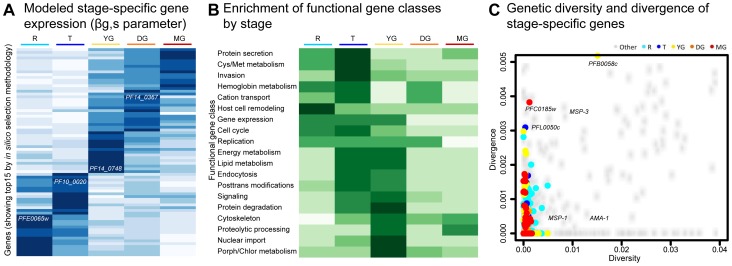
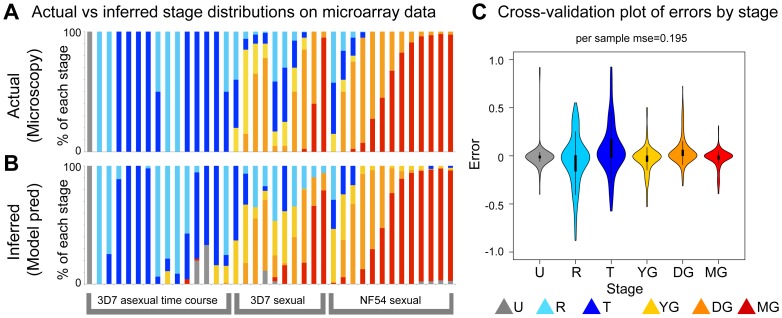
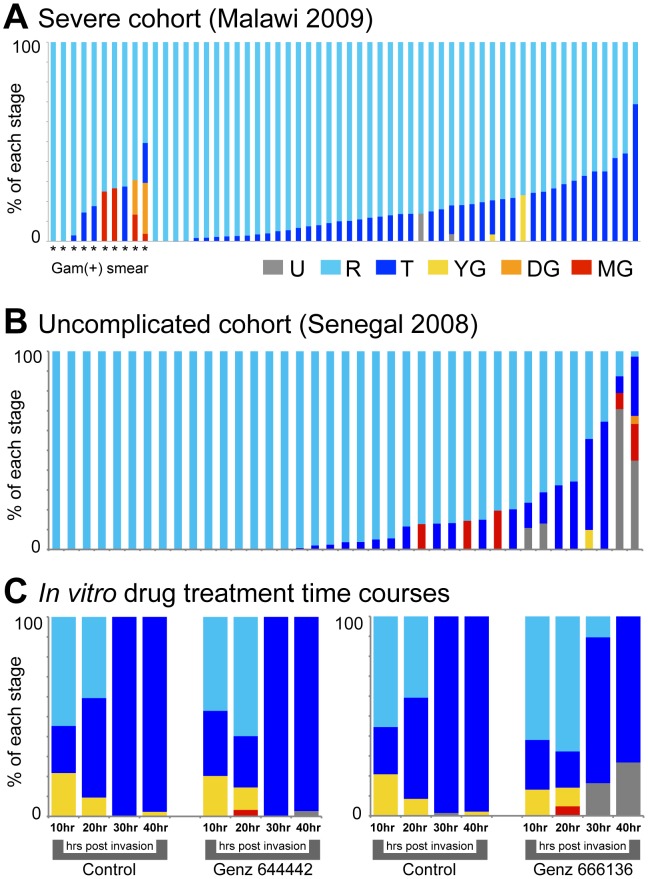
In the current era of malaria eradication, reducing transmission is critical. Assessment of transmissibility requires tools that can accurately identify the various developmental stages of the malaria parasite, particularly those required for transmission (sexual stages). Here, we present a method for estimating relative amounts of Plasmodium falciparum asexual and sexual stages from gene expression measurements. These are modeled using constrained linear regression to characterize stage-specific expression profiles within mixed-stage populations. The resulting profiles were analyzed functionally by gene set enrichment analysis (GSEA), confirming differentially active pathways such as increased mitochondrial activity and lipid metabolism during sexual development. We validated model predictions both from microarrays and from quantitative RT-PCR (qRT-PCR) measurements, based on the expression of a small set of key transcriptional markers. This sufficient marker set was identified by backward selection from the whole genome as available from expression arrays, targeting one sentinel marker per stage. The model as learned can be applied to any new microarray or qRT-PCR transcriptional measurement. We illustrate its use in vitro in inferring changes in stage distribution following stress and drug treatment and in vivo in identifying immature and mature sexual stage carriers within patient cohorts. We believe this approach will be a valuable resource for staging lab and field samples alike and will have wide applicability in epidemiological studies of malaria transmission.
在当前消除疟疾的时代,降低传播至关重要。评估传染性需要能够准确识别疟原虫各个发育阶段的工具,特别是那些用于传播的阶段(有性阶段)。在这里,我们提出了一种从基因表达测量中估计疟原虫无性和有性阶段相对数量的方法。这些方法使用约束线性回归进行建模,以描述混合阶段群体中的特定阶段表达谱。通过基因集富集分析(GSEA)对得到的表达谱进行功能分析,证实了在有性发育过程中增加线粒体活性和脂质代谢等差异活跃的途径。我们基于一小部分关键转录标志物的表达,通过从微阵列和定量 RT-PCR(qRT-PCR)测量中验证了模型预测。通过从表达阵列中可用的全基因组向后选择来确定这个足够的标志物集,每个阶段都有一个有代表性的标志物。所学习的模型可以应用于任何新的微阵列或 qRT-PCR 转录测量。我们在体外演示了它在推断应激和药物治疗后阶段分布变化以及在体内识别患者队列中未成熟和成熟有性阶段携带者方面的应用。我们相信,这种方法将成为实验室和现场样本分期的有价值资源,并且在疟疾传播的流行病学研究中具有广泛的适用性。