College of Life Science, State Key Laboratory of Crop Stress Biology for Arid Areas, Northwest A&F University, Yangling, Shaanxi, China.
BMC Genomics. 2013 Dec 21;14:910. doi: 10.1186/1471-2164-14-910.
Determination of the minimum gene set for cellular life is one of the central goals in biology. Genome-wide essential gene identification has progressed rapidly in certain bacterial species; however, it remains difficult to achieve in most eukaryotic species. Several computational models have recently been developed to integrate gene features and used as alternatives to transfer gene essentiality annotations between organisms.
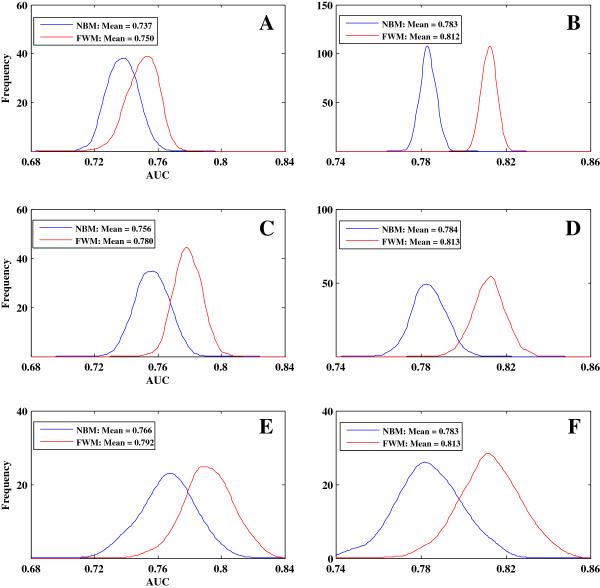
We first collected features that were widely used by previous predictive models and assessed the relationships between gene features and gene essentiality using a stepwise regression model. We found two issues that could significantly reduce model accuracy: (i) the effect of multicollinearity among gene features and (ii) the diverse and even contrasting correlations between gene features and gene essentiality existing within and among different species. To address these issues, we developed a novel model called feature-based weighted Naïve Bayes model (FWM), which is based on Naïve Bayes classifiers, logistic regression, and genetic algorithm. The proposed model assesses features and filters out the effects of multicollinearity and diversity. The performance of FWM was compared with other popular models, such as support vector machine, Naïve Bayes model, and logistic regression model, by applying FWM to reciprocally predict essential genes among and within 21 species. Our results showed that FWM significantly improves the accuracy and robustness of essential gene prediction.
FWM can remarkably improve the accuracy of essential gene prediction and may be used as an alternative method for other classification work. This method can contribute substantially to the knowledge of the minimum gene sets required for living organisms and the discovery of new drug targets.
确定细胞生命所需的最小基因集是生物学的核心目标之一。在某些细菌物种中,全基因组必需基因的鉴定已经取得了快速进展;然而,在大多数真核生物物种中,这仍然是困难的。最近已经开发了几种计算模型来整合基因特征,并用作在生物体之间转移基因必需性注释的替代方法。
我们首先收集了以前预测模型广泛使用的特征,并使用逐步回归模型评估了基因特征与基因必需性之间的关系。我们发现了两个可能显著降低模型准确性的问题:(i)基因特征之间的多线性影响,以及(ii)不同物种内部和之间基因特征与基因必需性之间存在的多样甚至相反的相关性。为了解决这些问题,我们开发了一种称为基于特征的加权朴素贝叶斯模型(FWM)的新模型,该模型基于朴素贝叶斯分类器、逻辑回归和遗传算法。所提出的模型评估特征并过滤掉多线性和多样性的影响。通过将 FWM 应用于 21 个物种之间和内部的必需基因相互预测,将 FWM 的性能与其他流行模型(如支持向量机、朴素贝叶斯模型和逻辑回归模型)进行了比较。我们的结果表明,FWM 显著提高了必需基因预测的准确性和稳健性。
FWM 可以显著提高必需基因预测的准确性,并且可以用作其他分类工作的替代方法。这种方法可以为生物体所需的最小基因集的知识和新药物靶点的发现做出重要贡献。