Bioinformatics Unit, Microbiology Services (Colindale), Public Health England, 61 Colindale Avenue, London, NW9 5EQ, UK.
BMC Microbiol. 2013 Dec 24;13:302. doi: 10.1186/1471-2180-13-302.
Legionella pneumophila is an opportunistic pathogen of humans where the source of infection is usually from contaminated man-made water systems. When an outbreak of Legionnaires' disease caused by L. pneumophila occurs, it is necessary to discover the source of infection. A seven allele sequence-based typing scheme (SBT) has been very successful in providing the means to attribute outbreaks of L. pneumophila to a particular source or sources. Particular sequence types described by this scheme are known to exhibit specific phenotypes. For instance some types are seen often in clinical cases but are rarely isolated from the environment and vice versa. Of those causing human disease some types are thought to be more likely to cause more severe disease. It is possible that the genetic basis for these differences are vertically inherited and associated with particular genetic lineages within the population. In order to provide a framework within which to test this hypothesis and others relating to the population biology of L. pneumophila, a set of genomes covering the known diversity of the organism is required.
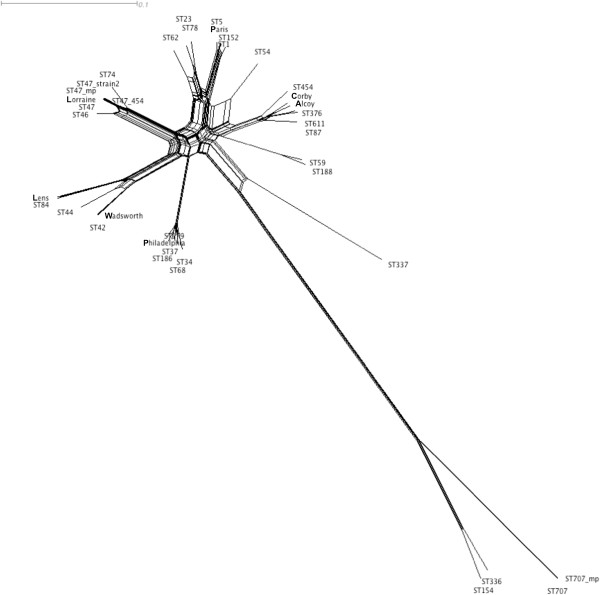
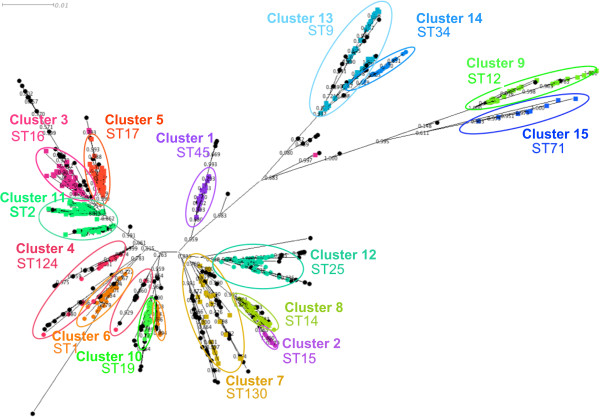
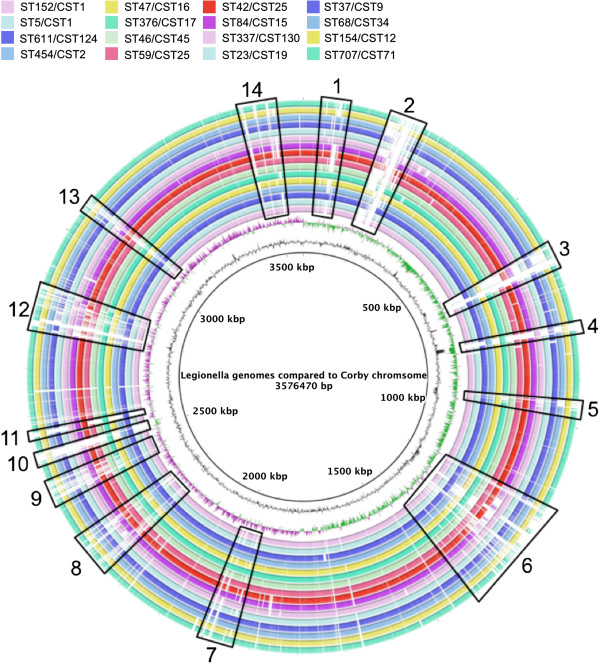
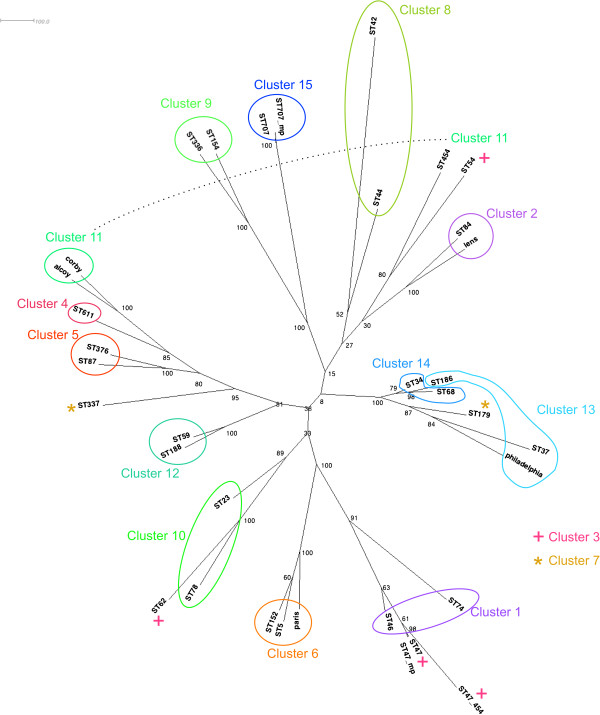
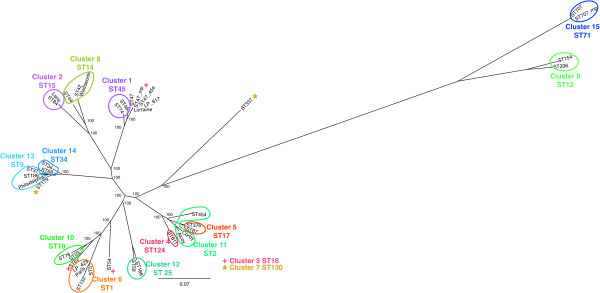
Firstly, this study describes a means to group L. pneumophila strains into pragmatic clusters, using a methodology that takes into consideration the genetic forces operating on the population. These clusters can be used as a standardised nomenclature, so those wishing to describe a group of strains can do so. Secondly, the clusters generated from the first part of the study were used to select strains rationally for whole genome sequencing (WGS). The data generated was used to compare phylogenies derived from SBT and WGS. In general the SBT sequence type (ST) accurately reflects the whole genome-based genotype. Where there are exceptions and recombination has resulted in the ST no longer reflecting the genetic lineage described by the whole genome sequence, the clustering technique employed detects these sequence types as being admixed, indicating their mixed inheritance.
We conclude that SBT is usually a good proxy for the genetic lineage described by the whole genome, and therefore utility of SBT is still suitable until the technology and economics of high throughput sequencing reach the point where routine WGS of L. pneumophila isolates for outbreak investigation is feasible.
嗜肺军团菌是人类的机会性病原体,感染源通常来自受污染的人造水系统。当发生由嗜肺军团菌引起的军团病爆发时,有必要发现感染源。一种基于七个等位基因序列的分型方案(SBT)非常成功地提供了将嗜肺军团菌的爆发归因于特定来源或来源的方法。该方案描述的特定序列类型已知表现出特定的表型。例如,某些类型经常在临床病例中出现,但很少从环境中分离出来,反之亦然。引起人类疾病的某些类型被认为更有可能引起更严重的疾病。这些差异的遗传基础可能是垂直遗传的,并与种群内的特定遗传谱系相关。为了提供一个框架来检验这一假设和其他与嗜肺军团菌种群生物学有关的假设,需要一组涵盖该生物体已知多样性的基因组。
首先,本研究描述了一种使用考虑到影响种群的遗传力的方法将嗜肺军团菌菌株分为实用聚类的方法。这些聚类可以用作标准化命名法,因此那些希望描述一组菌株的人可以这样做。其次,从研究的第一部分生成的聚类用于合理选择用于全基因组测序(WGS)的菌株。生成的数据用于比较来自 SBT 和 WGS 的系统发育。一般来说,SBT 序列型(ST)准确反映了基于全基因组的基因型。在 ST 不再反映全基因组序列描述的遗传谱系的情况下,由于重组导致的例外情况,使用的聚类技术检测到这些序列类型是混合的,表明它们的混合遗传。
我们的结论是,SBT 通常是全基因组描述的遗传谱系的良好替代物,因此,在高通量测序的技术和经济达到常规进行嗜肺军团菌分离物爆发调查的全基因组测序的可行水平之前,SBT 的实用性仍然适用。