Department of Pathology, Case Western Reserve University, Cleveland, Ohio, United States of America ; Division of Infectious Diseases and HIV Medicine, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, United States of America.
Division of Infectious Diseases and HIV Medicine, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, United States of America.
PLoS Pathog. 2013;9(12):e1003809. doi: 10.1371/journal.ppat.1003809. Epub 2013 Dec 19.
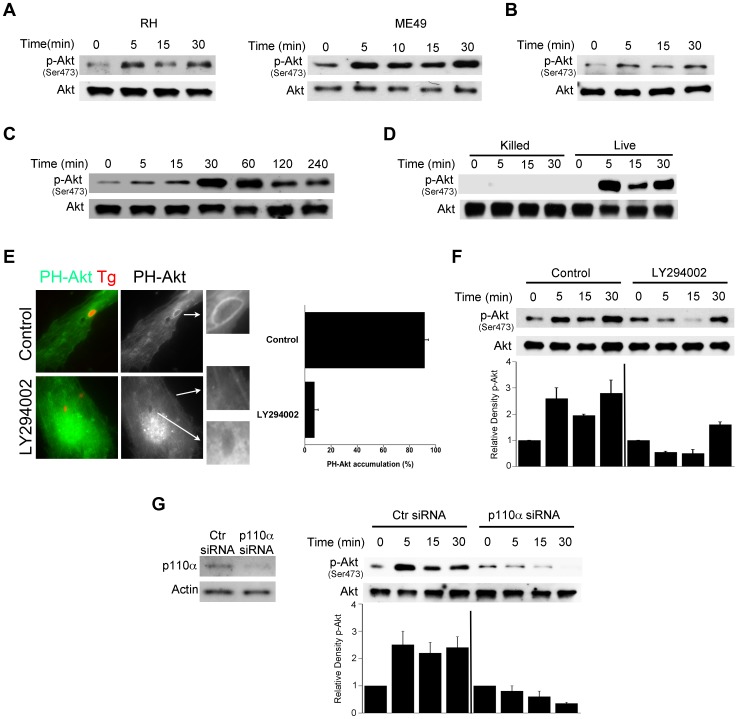
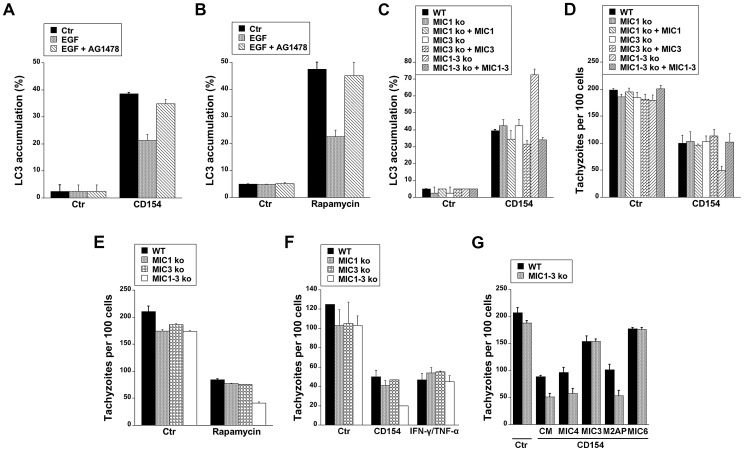
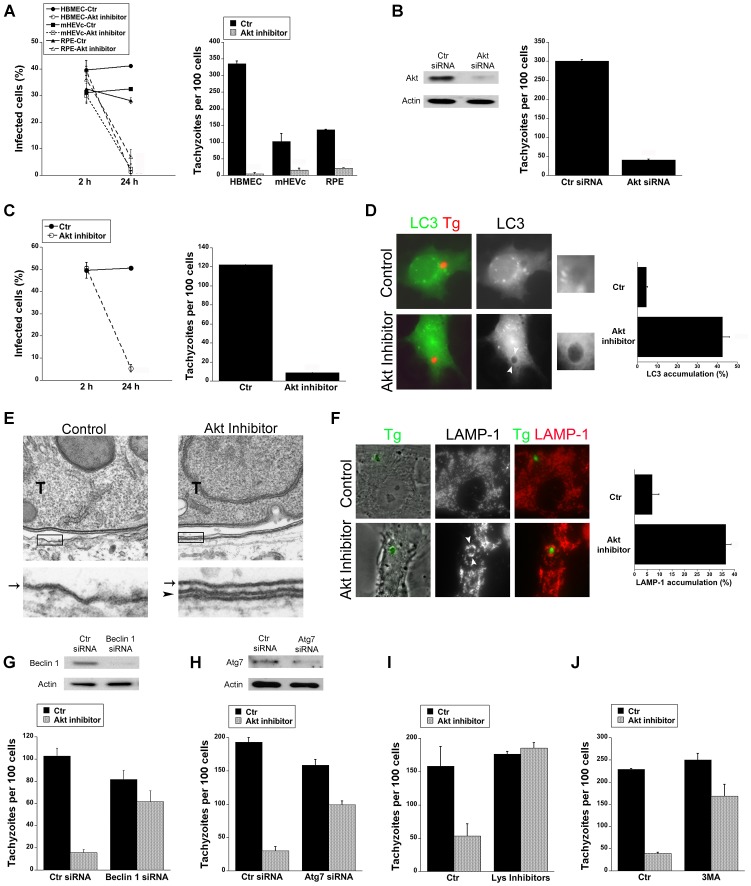
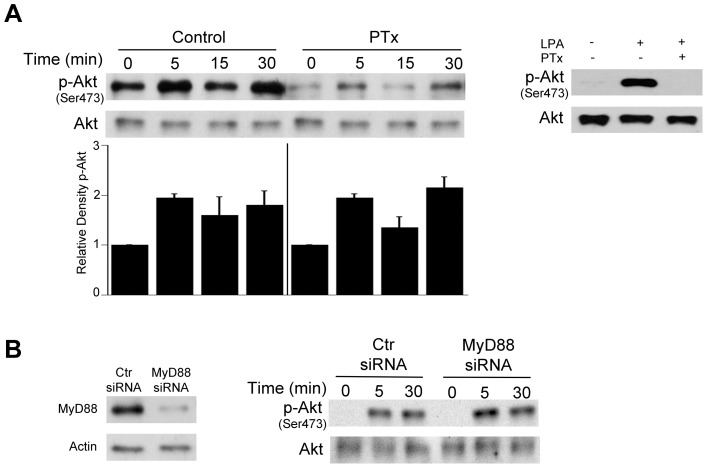
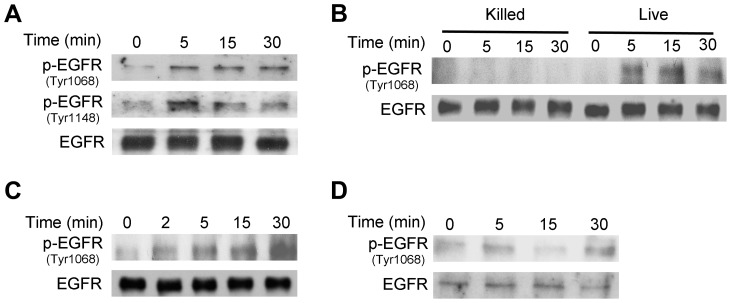
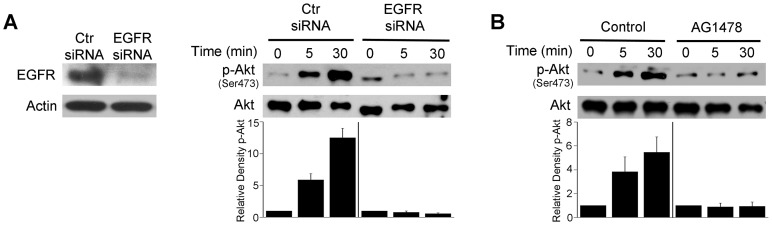
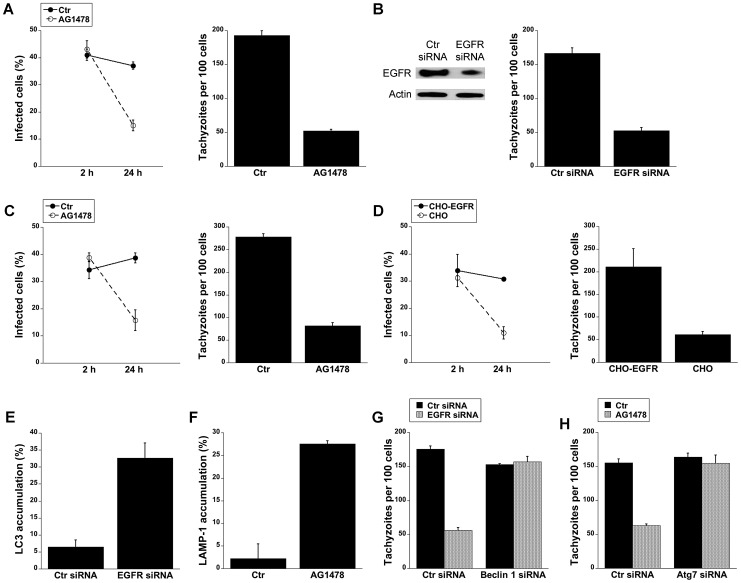
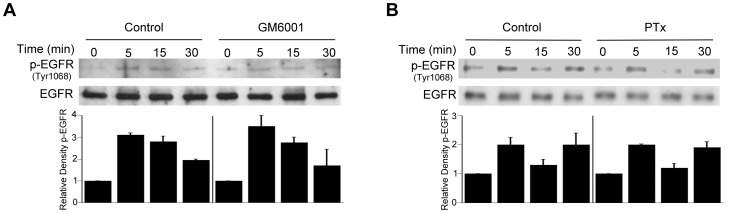
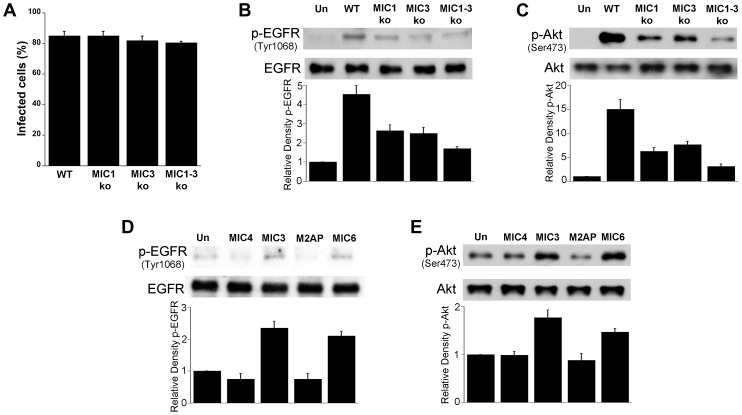
Toxoplasma gondii resides in an intracellular compartment (parasitophorous vacuole) that excludes transmembrane molecules required for endosome-lysosome recruitment. Thus, the parasite survives by avoiding lysosomal degradation. However, autophagy can re-route the parasitophorous vacuole to the lysosomes and cause parasite killing. This raises the possibility that T. gondii may deploy a strategy to prevent autophagic targeting to maintain the non-fusogenic nature of the vacuole. We report that T. gondii activated EGFR in endothelial cells, retinal pigment epithelial cells and microglia. Blockade of EGFR or its downstream molecule, Akt, caused targeting of the parasite by LC3(+) structures, vacuole-lysosomal fusion, lysosomal degradation and killing of the parasite that were dependent on the autophagy proteins Atg7 and Beclin 1. Disassembly of GPCR or inhibition of metalloproteinases did not prevent EGFR-Akt activation. T. gondii micronemal proteins (MICs) containing EGF domains (EGF-MICs; MIC3 and MIC6) appeared to promote EGFR activation. Parasites defective in EGF-MICs (MIC1 ko, deficient in MIC1 and secretion of MIC6; MIC3 ko, deficient in MIC3; and MIC1-3 ko, deficient in MIC1, MIC3 and secretion of MIC6) caused impaired EGFR-Akt activation and recombinant EGF-MICs (MIC3 and MIC6) caused EGFR-Akt activation. In cells treated with autophagy stimulators (CD154, rapamycin) EGFR signaling inhibited LC3 accumulation around the parasite. Moreover, increased LC3 accumulation and parasite killing were noted in CD154-activated cells infected with MIC1-3 ko parasites. Finally, recombinant MIC3 and MIC6 inhibited parasite killing triggered by CD154 particularly against MIC1-3 ko parasites. Thus, our findings identified EGFR activation as a strategy used by T. gondii to maintain the non-fusogenic nature of the parasitophorous vacuole and suggest that EGF-MICs have a novel role in affecting signaling in host cells to promote parasite survival.
刚地弓形虫位于细胞内隔室(滋养体空泡)中,该隔室排斥招募内体-溶酶体所需的跨膜分子。因此,寄生虫通过避免溶酶体降解而存活。然而,自噬可以将滋养体空泡重新路由到溶酶体并导致寄生虫死亡。这就提出了一种可能性,即刚地弓形虫可能会部署一种策略来防止自噬靶向,以维持空泡的非融合性质。我们报告说,刚地弓形虫在血管内皮细胞、视网膜色素上皮细胞和小胶质细胞中激活了 EGFR。EGFR 或其下游分子 Akt 的阻断导致寄生虫被 LC3(+)结构靶向,空泡-溶酶体融合,溶酶体降解,寄生虫被杀死,这依赖于自噬蛋白 Atg7 和 Beclin 1。GPCR 的解体或金属蛋白酶的抑制并不能阻止 EGFR-Akt 的激活。含有 EGF 结构域的刚地弓形虫微线蛋白(MIC)(EGF-MIC;MIC3 和 MIC6)似乎促进了 EGFR 的激活。缺乏 EGF-MIC(MIC1 ko,MIC1 和 MIC6 分泌缺陷;MIC3 ko,MIC3 缺陷;MIC1-3 ko,MIC1、MIC3 和 MIC6 分泌缺陷)的寄生虫导致 EGFR-Akt 激活受损,重组 EGF-MIC(MIC3 和 MIC6)导致 EGFR-Akt 激活。在用自噬刺激物(CD154、雷帕霉素)处理的细胞中,EGFR 信号抑制了寄生虫周围 LC3 的积累。此外,在感染 MIC1-3 ko 寄生虫的 CD154 激活细胞中,观察到 LC3 积累增加和寄生虫杀伤增加。最后,重组 MIC3 和 MIC6 抑制了 CD154 触发的寄生虫杀伤,尤其是针对 MIC1-3 ko 寄生虫。因此,我们的研究结果确定 EGFR 激活是刚地弓形虫维持滋养体空泡非融合性质的一种策略,并表明 EGF-MIC 在影响宿主细胞信号以促进寄生虫存活方面具有新的作用。