Voskoboinik Ilia, Trapani Joseph A
Killer Cell Biology Laboratory, Peter MacCallum Cancer Centre , East Melbourne, VIC , Australia ; Sir Peter MacCallum Department of Oncology, The University of Melbourne , Melbourne, VIC , Australia.
Sir Peter MacCallum Department of Oncology, The University of Melbourne , Melbourne, VIC , Australia ; Cancer Cell Death Laboratory, Peter MacCallum Cancer Centre , East Melbourne, VIC , Australia.
Front Immunol. 2013 Dec 12;4:441. doi: 10.3389/fimmu.2013.00441.
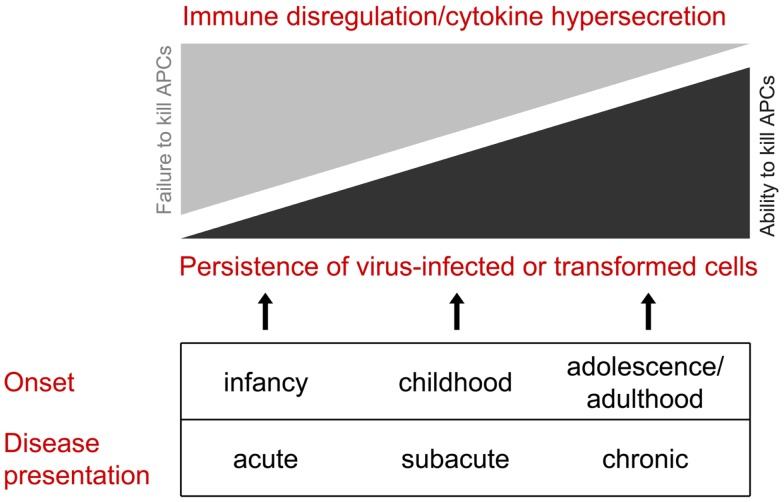
Congenital perforin deficiency is considered a rare cause of human immunopathology and immune dysregulation, and classically presents as a fatal illness early in infancy. However, we propose that a group of related disorders in which killer lymphocytes deliver only partially active perforin or a reduced quantum of wild-type perforin to the immune synapse should be considered part of an extended syndrome with overlapping but more variable clinical features. Apart from the many rare mutations scattered over the coding sequences, up to 10% of Caucasians carry the severely hypomorphic PRF1 allele C272 > T (leading to A91V mutation) and the overall prevalence of the homozygous state for A91V is around 1 in 600 individuals. We therefore postulate that the partial loss of perforin function and its clinical consequences may be more common then currently suspected. An acute clinical presentation is infrequent in A91V heterozygous individuals, but we postulate that the partial loss of perforin function may potentially be manifested in childhood or early adulthood as "idiopathic" inflammatory disease, or through increased cancer susceptibility - either hematological malignancy or multiple, independent primary cancers. We suggest the new term "perforinopathy" to signify the common functional endpoints of all the known consequences of perforin deficiency and failure to deliver fully functional perforin.
先天性穿孔素缺乏被认为是人类免疫病理学和免疫失调的罕见原因,经典表现为婴儿早期的致命疾病。然而,我们提出,一组相关疾病,其中杀伤淋巴细胞仅向免疫突触传递部分活性穿孔素或减少量的野生型穿孔素,应被视为具有重叠但更具变异性临床特征的扩展综合征的一部分。除了散布在编码序列中的许多罕见突变外,高达10%的高加索人携带严重低功能的PRF1等位基因C272>T(导致A91V突变),A91V纯合状态的总体患病率约为600人中1人。因此,我们推测穿孔素功能的部分丧失及其临床后果可能比目前怀疑的更为常见。A91V杂合个体很少出现急性临床表现,但我们推测穿孔素功能的部分丧失可能在儿童期或成年早期表现为“特发性”炎症性疾病,或通过增加癌症易感性——血液系统恶性肿瘤或多种独立的原发性癌症。我们建议使用新术语“穿孔素病”来表示穿孔素缺乏和未能传递完全功能性穿孔素的所有已知后果的共同功能终点。