Terzuoli Erika, Meini Stefania, Cucchi Paola, Catalani Claudio, Cialdai Cecilia, Maggi Carlo Alberto, Giachetti Antonio, Ziche Marina, Donnini Sandra
Department of Life Sciences, University of Siena, Siena, Italy¸
Pharmacology Department, Menarini Ricerche S.p.A, Florence, Italy.
PLoS One. 2014 Jan 2;9(1):e84358. doi: 10.1371/journal.pone.0084358. eCollection 2014.
Bradykinin (BK) induces angiogenesis by promoting vessel permeability, growth and remodeling. This study aimed to demonstrate that the B2R antagonist, fasitibant, inhibits the BK pro-angiogenic effects.
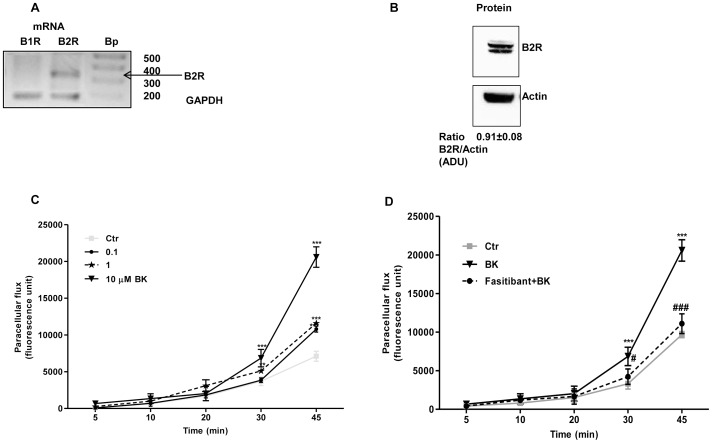
We assesed the ability of fasibitant to antagonize the BK stimulation of cultured human cells (HUVEC) and circulating pro-angiogenic cells (PACs), in producing cell permeability (paracellular flux), migration and pseocapillary formation. The latter parameter was studied in vitro (matrigel assay) and in vivo in mice (matrigel plug) and in rat model of experimental osteoarthritis (OA). We also evaluated NF-κB activation in cultured cells by measuring its nuclear translocation and its downstream effectors such as the proangiogenic ciclooxygenase-2 (COX-2), prostaglandin E-2 and vascular endothelial growth factor (VEGF).
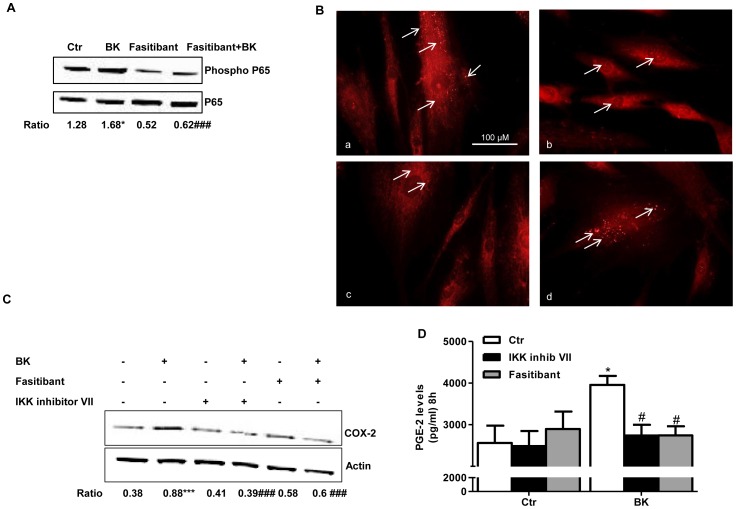
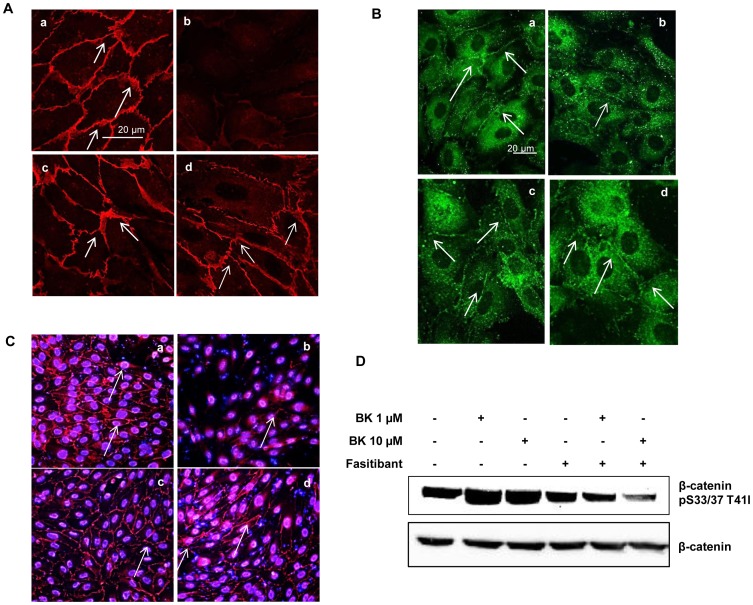
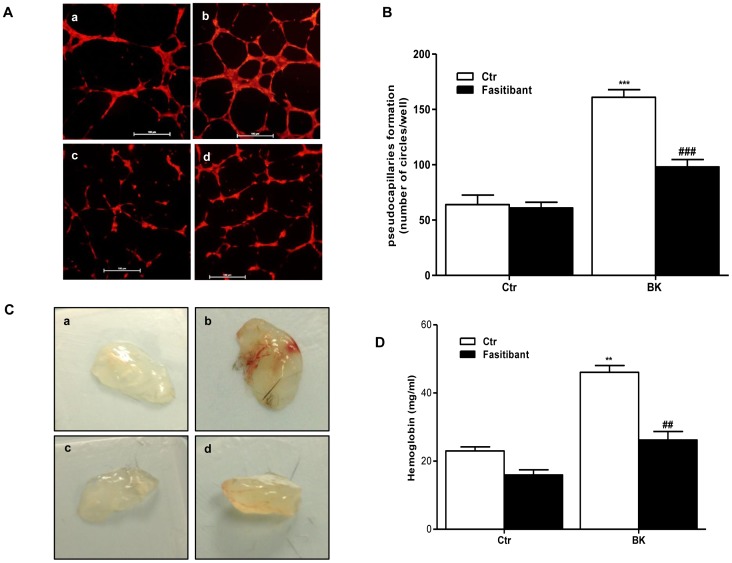
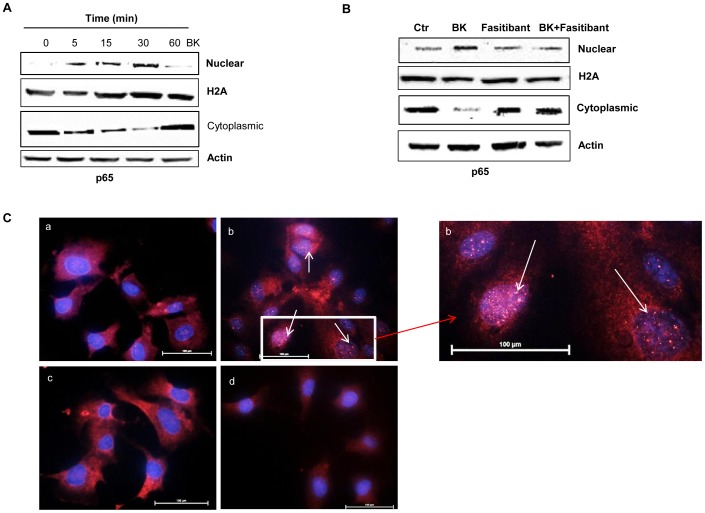
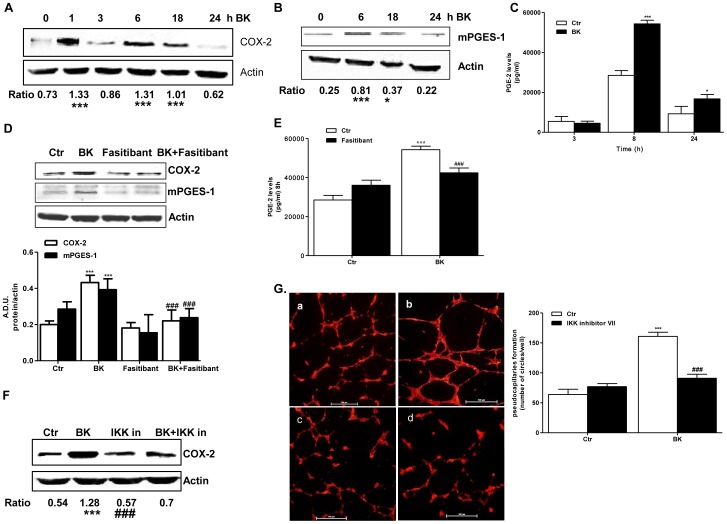
HUVEC, exposed to BK (1-10 µM), showed increased permeability, disassembly of adherens and tight-junction, increased cell migration, and pseudocapillaries formation. We observed a significant increase of vessel density in the matrigel assay in mice and in rats OA model. Importantly, B2R stimulation elicited, both in HUVEC and PACs, NF-κB activation, leading to COX-2 overexpression, enhanced prostaglandin E-2 production. and VEGF output. The BK/NF-κB axis, and the ensuing amplification of inflammatory/angiogenic responses were fully prevented by fasitibant as well as by IKK VII, an NF-κB. Inhibitor.
This work illustrates the role of the endothelium in the inflammation provoked by the BK/NF-κB axis. It also demonstates that B2R blockade by the antaogonist fasibitant, abolishes both the initial stimulus and its amplification, strongly attenuating the propagation of inflammation.
缓激肽(BK)通过促进血管通透性、生长和重塑来诱导血管生成。本研究旨在证明B2R拮抗剂法西替班可抑制BK的促血管生成作用。
我们评估了法西替班拮抗BK刺激培养的人细胞(HUVEC)和循环促血管生成细胞(PACs)产生细胞通透性(细胞旁通量)、迁移和伪毛细血管形成的能力。后一参数在体外(基质胶试验)以及在小鼠体内(基质胶栓)和实验性骨关节炎(OA)大鼠模型中进行了研究。我们还通过测量其核转位及其下游效应分子,如促血管生成的环氧化酶-2(COX-2)、前列腺素E-2和血管内皮生长因子(VEGF),评估了培养细胞中NF-κB的激活情况。
暴露于BK(1-10μM)的HUVEC显示通透性增加、黏附连接和紧密连接解体、细胞迁移增加以及伪毛细血管形成。我们在小鼠和大鼠OA模型的基质胶试验中观察到血管密度显著增加。重要的是,B2R刺激在HUVEC和PACs中均引发NF-κB激活,导致COX-2过表达、前列腺素E-2产生增加以及VEGF分泌增加。法西替班以及NF-κB抑制剂IKK VII完全阻断了BK/NF-κB轴以及随之而来的炎症/血管生成反应的放大。
这项工作阐明了内皮细胞在BK/NF-κB轴引发的炎症中的作用。它还表明拮抗剂法西替班阻断B2R可消除初始刺激及其放大作用,强烈减弱炎症的传播。