Department of Neurology and Neurosurgery, McGill University, Montreal, QC H3A 2B4, Canada.
Acta Neuropathol Commun. 2014 Jan 8;2:5. doi: 10.1186/2051-5960-2-5.
Spontaneous autoimmune peripheral neuropathy including Guillain-Barré Syndrome (GBS) represents as one of the serious emergencies in neurology. Although pathological changes have been well documented, molecular and cellular mechanisms of GBS are still under-explored, partially due to short of appropriate animal models. The field lacks of spontaneous and translatable models for mechanistic investigations. As GBS is preceded often by viral or bacterial infection, a condition can enhance co-stimulatory activity; we sought to investigate the critical role of T cell co-stimulation in this autoimmune disease.
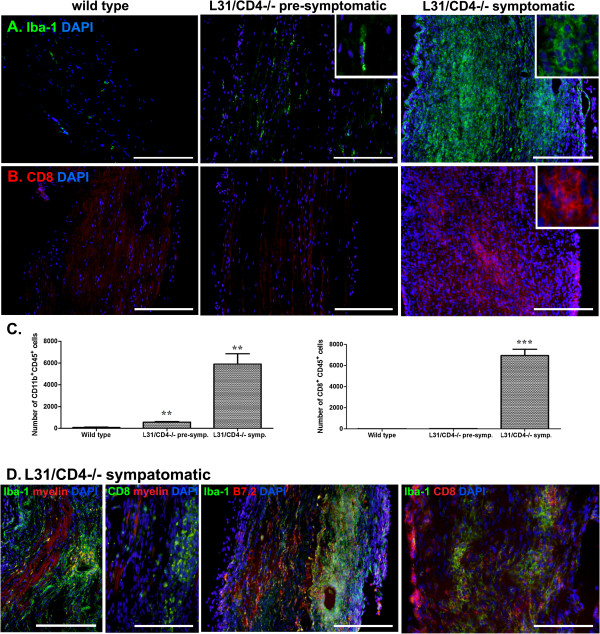
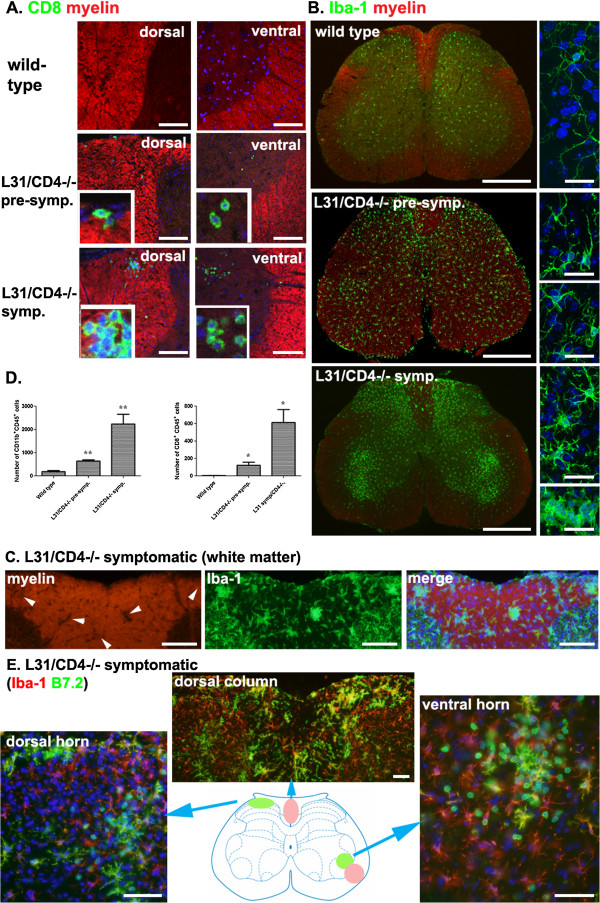
Our previous study reported that transgene-derived constitutive expression of co-stimulator B7.2 on antigen presenting cells of the nervous tissues drove spontaneous neurological disorders. Depletion of CD4+ T cells in L31 mice accelerated the onset and increased the prevalence of the disease. In the current study, we further demonstrated that L31/CD4-/- mice exhibited both motor and sensory deficits, including weakness and paresis of limbs, numbness to mechanical stimuli and hypersensitivity to thermal stimulation. Pathological changes were characterized by massive infiltration of macrophages and CD8+ T cells, demyelination and axonal damage in peripheral nerves, while changes in spinal cords could be secondary to the PNS damage. In symptomatic L31/CD4-/- mice, the disruption of the blood neural barriers was observed mainly in peripheral nerves. Interestingly, the infiltration of immune cells was initiated in pre-symptomatic L31/CD4-/- mice, prior to the disease onset, in the DRG and spinal roots where the blood nerve barrier is virtually absent.
L31/CD4-/- mice mimic most parts of clinical and pathological signatures of GBS in human; thus providing an unconventional opportunity to experimentally explore the critical events that lead to spontaneous, autoimmune demyelinating disease of the peripheral nervous system.
自发性自身免疫性周围神经病,包括格林-巴利综合征(GBS),是神经病学中的一种严重急症。尽管其病理变化已有详细记录,但 GBS 的分子和细胞机制仍未得到充分探索,部分原因是缺乏合适的动物模型。该领域缺乏用于机制研究的自发性和可转化的模型。由于 GBS 通常先于病毒或细菌感染,这种情况会增强共刺激活性;因此,我们试图研究 T 细胞共刺激在这种自身免疫性疾病中的关键作用。
我们之前的研究报告称,在神经组织的抗原呈递细胞上,转基因组成型表达共刺激分子 B7.2 会导致自发性神经紊乱。在 L31 小鼠中耗尽 CD4+T 细胞会加速疾病的发作并增加其患病率。在本研究中,我们进一步证明 L31/CD4-/- 小鼠表现出运动和感觉功能障碍,包括四肢无力和瘫痪、对机械刺激的麻木和对热刺激的过敏。病理变化的特征是大量巨噬细胞和 CD8+T 细胞浸润、周围神经脱髓鞘和轴突损伤,而脊髓的变化可能是 PNS 损伤的继发反应。在有症状的 L31/CD4-/- 小鼠中,主要在外周神经中观察到血神经屏障的破坏。有趣的是,免疫细胞的浸润始于疾病发作前的有症状 L31/CD4-/- 小鼠的背根神经节和脊神经根,在那里血神经屏障几乎不存在。
L31/CD4-/- 小鼠模拟了人类 GBS 的大部分临床和病理特征;因此,为实验探索导致自发性、自身免疫性周围神经系统脱髓鞘疾病的关键事件提供了一个非传统的机会。