Vandal Guillaume, Geiling Benjamin, Dankort David
Department of Biology, McGill University, Montréal, Quebec, Canada.
PLoS One. 2014 Jan 28;9(1):e84745. doi: 10.1371/journal.pone.0084745. eCollection 2014.
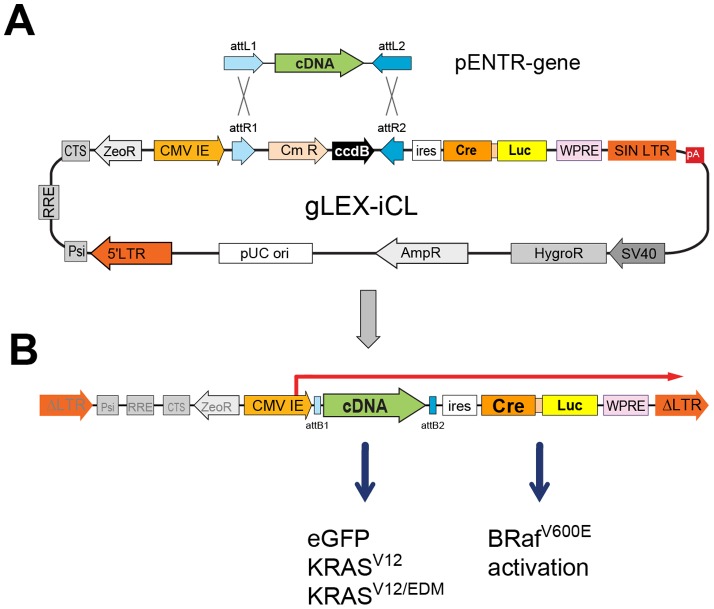
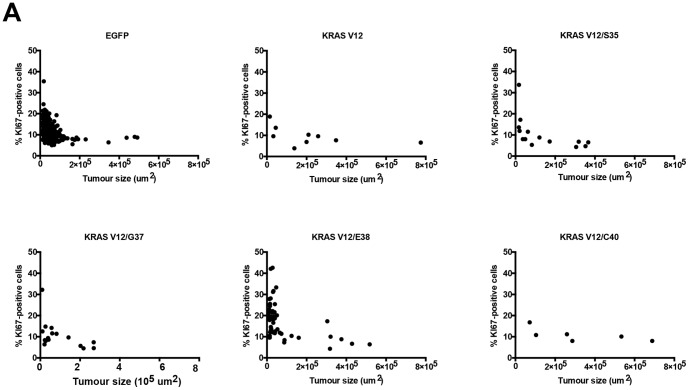
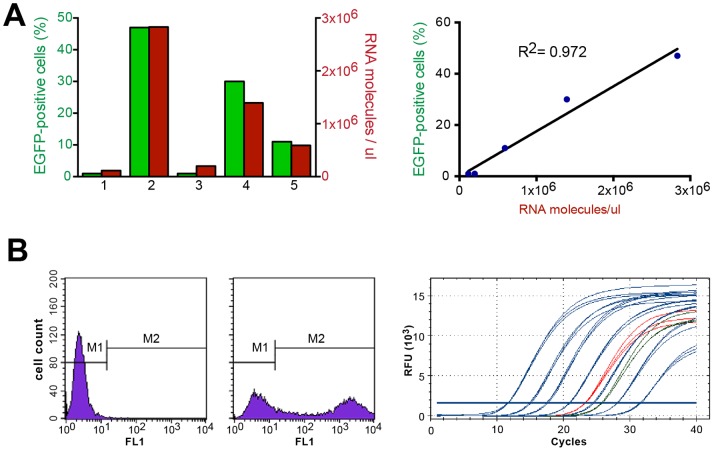
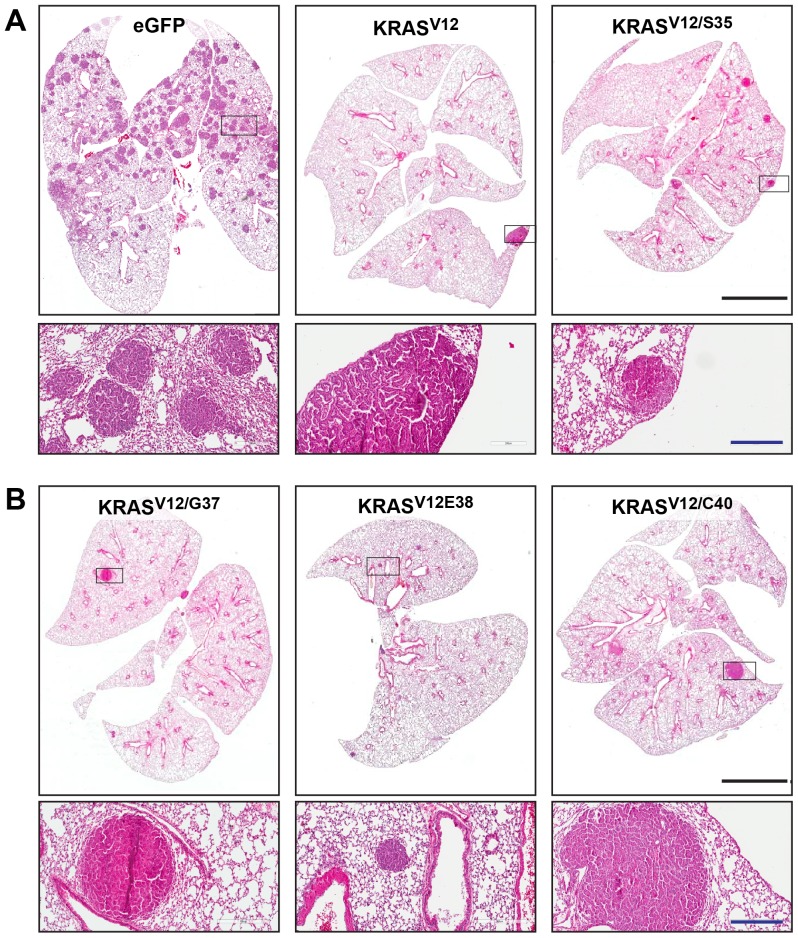
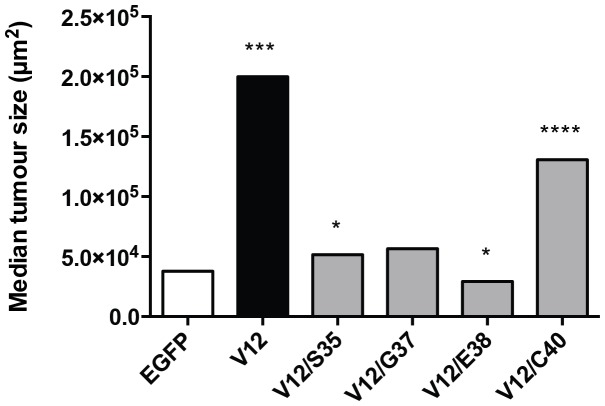
Lung cancer is currently the most deadly malignancy in industrialized countries and accounts for 18% of all cancer-related deaths worldwide. Over 70% of patients with non-small cell lung cancer (NSCLC) are diagnosed at a late stage, with a 5-year survival below 10%. KRAS and the EGFR are frequently mutated in NSCLC and while targeted therapies for patients with EGFR mutations exist, oncogenic KRAS is thus far not druggable. KRAS activates multiple signalling pathways, including the PI3K/Akt pathway, the Raf-Mek-Erk pathway and the RalGDS/Ral pathway. Lung-specific expression of BrafV600E, the most prevalent BRAF mutation found in human tumors, results in Raf-Mek-Erk pathway activation and in the formation of benign adenomas that undergo widespread senescence in a Cre-activated Braf mouse model (Braf(CA)). However, oncogenic KRAS expression in mice induces adenocarcinomas, suggesting additional KRAS-activated pathways cooperate with sustained RAF-MEK-ERK signalling to bypass the oncogene-induced senescence proliferation arrest. To determine which KRAS effectors were responsible for tumor progression, we created four effector domain mutants (S35, G37, E38 and C40) in G12V-activated KRAS and expressed these alone or with BrafV600E in mouse lungs... The S35 and E38 mutants bind to Raf proteins but not PI3K or RalGDS; the G37 mutant binds to RalGDS and not Raf or PI3K and the C40 mutant is specific to PI3K. We designed lentiviral vectors to code for Cre recombinase along with KRAS mutants (V12, V12/S35, V12/G37, V12/E38 or V12/C40) or EGFP as a negative control.. These lentiviruses were used to infect Braf(CA) and wild-type mice. Surprisingly there was a significant decrease in tumor number and penetrance with each KRAS effector domain mutant relative to controls, suggesting that KRAS directly activates effectors with tumor suppressive functions.
肺癌是目前工业化国家中最致命的恶性肿瘤,占全球所有癌症相关死亡人数的18%。超过70%的非小细胞肺癌(NSCLC)患者在晚期被诊断出来,其5年生存率低于10%。KRAS和表皮生长因子受体(EGFR)在NSCLC中经常发生突变,虽然存在针对EGFR突变患者的靶向治疗,但致癌性KRAS迄今为止仍无药可治。KRAS激活多种信号通路,包括磷脂酰肌醇-3激酶/蛋白激酶B(PI3K/Akt)通路、Raf-丝裂原活化蛋白激酶/细胞外信号调节激酶(Raf-Mek-Erk)通路和Ral鸟苷酸解离刺激因子/Ral(RalGDS/Ral)通路。在人类肿瘤中发现的最常见的BRAF突变BrafV600E在肺中的特异性表达,导致Raf-Mek-Erk通路激活,并在Cre激活的Braf小鼠模型(Braf(CA))中形成广泛衰老的良性腺瘤。然而,致癌性KRAS在小鼠中的表达诱导腺癌,这表明额外的KRAS激活通路与持续的RAF-MEK-ERK信号协同作用,以绕过癌基因诱导的衰老增殖停滞。为了确定哪些KRAS效应器负责肿瘤进展,我们在G12V激活的KRAS中创建了四个效应器结构域突变体(S35、G37、E38和C40),并在小鼠肺中单独或与BrafV600E一起表达这些突变体……S35和E38突变体与Raf蛋白结合,但不与PI3K或RalGDS结合;G37突变体与RalGDS结合,不与Raf或PI3K结合,C40突变体对PI3K具有特异性。我们设计了慢病毒载体,用于编码Cre重组酶以及KRAS突变体(V12、V12/S35、V12/G37、V12/E38或V12/C40)或作为阴性对照的增强型绿色荧光蛋白(EGFP)……这些慢病毒用于感染Braf(CA)和野生型小鼠。令人惊讶的是,相对于对照组,每个KRAS效应器结构域突变体的肿瘤数量和发生率都显著降低,这表明KRAS直接激活具有肿瘤抑制功能的效应器。